This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
proofread
To streamline drug discovery, team develops algorithmic framework to identify optimal molecular candidates
. DOI: 10.1038/s43588-024-00639-y")
The use of AI to streamline drug discovery is exploding. Researchers are deploying machine-learning models to help them identify molecules, among billions of options, that might have the properties they are seeking to develop new medicines.
But there are so many variables to consider—from the price of materials to the risk of something going wrong—that even when scientists use AI, weighing the costs of synthesizing the best candidates is no easy task.
The myriad challenges involved in identifying the best and most cost-efficient molecules to test is one reason new medicines take so long to develop, as well as a key driver of high prescription drug prices.
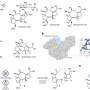
To help scientists make cost-aware choices, MIT researchers have developed an algorithmic framework to automatically identify optimal molecular candidates, which minimizes synthetic cost while maximizing the likelihood candidates have desired properties. The algorithm also identifies the materials and experimental steps needed to synthesize these molecules.
Their quantitative framework, known as Synthesis Planning and Rewards-based Route Optimization Workflow (SPARROW), considers the costs of synthesizing a batch of molecules at once, since multiple candidates can often be derived from some of the same chemical compounds. Moreover, this unified approach captures key information on molecular design, property prediction, and synthesis planning from online repositories and widely used AI tools.
The paper is published in the journal Nature Computational Science.
Beyond helping pharmaceutical companies discover new drugs more efficiently, SPARROW could be used in applications like the invention of new agrichemicals or the discovery of specialized materials for organic electronics.
"The selection of compounds is very much an art at the moment—and at times it is a very successful art. But because we have all these other models and predictive tools that give us information on how molecules might perform and how they might be synthesized, we can and should be using that information to guide the decisions we make," says Connor Coley, the Class of 1957 Career Development Assistant Professor in the MIT departments of Chemical Engineering and Electrical Engineering and Computer Science, and senior author of a paper on SPARROW.
Coley is joined on the paper by lead author Jenna Fromer.
Complex cost considerations
In a sense, whether a scientist should synthesize and test a certain molecule boils down to a question of the synthetic cost versus the value of the experiment. However, determining cost or value are tough problems on their own.
For instance, an experiment might require expensive materials or it could have a high risk of failure. On the value side, one might consider how useful it would be to know the properties of this molecule or whether those predictions carry a high level of uncertainty.
At the same time, pharmaceutical companies increasingly use batch synthesis to improve efficiency. Instead of testing molecules one at a time, they use combinations of chemical building blocks to test multiple candidates at once. However, this means the chemical reactions must all require the same experimental conditions. This makes estimating cost and value even more challenging.
SPARROW tackles this challenge by considering the shared intermediary compounds involved in synthesizing molecules and incorporating that information into its cost-versus-value function.
"When you think about this optimization game of designing a batch of molecules, the cost of adding on a new structure depends on the molecules you have already chosen," Coley says.
The framework also considers things like the costs of starting materials, the number of reactions that are involved in each synthetic route, and the likelihood those reactions will be successful on the first try.
To utilize SPARROW, a scientist provides a set of molecular compounds they are thinking of testing and a definition of the properties they are hoping to find.
From there, SPARROW collects information on the molecules and their synthetic pathways and then weighs the value of each one against the cost of synthesizing a batch of candidates. It automatically selects the best subset of candidates that meet the user's criteria and finds the most cost-effective synthetic routes for those compounds.
"It does all this optimization in one step, so it can really capture all of these competing objectives simultaneously," Fromer says.
A versatile framework
SPARROW is unique because it can incorporate molecular structures that have been hand-designed by humans, those that exist in virtual catalogs, or never-before-seen molecules that have been invented by generative AI models.
"We have all these different sources of ideas. Part of the appeal of SPARROW is that you can take all these ideas and put them on a level playing field," Coley adds.
The researchers evaluated SPARROW by applying it in three case studies. The case studies, based on real-world problems faced by chemists, were designed to test SPARROW's ability to find cost-efficient synthesis plans while working with a wide range of input molecules.
They found that SPARROW effectively captured the marginal costs of batch synthesis and identified common experimental steps and intermediate chemicals. In addition, it could scale up to handle hundreds of potential molecular candidates.
"In the machine-learning-for-chemistry community, there are so many models that work well for retrosynthesis or molecular property prediction, for example, but how do we actually use them? Our framework aims to bring out the value of this prior work. By creating SPARROW, hopefully we can guide other researchers to think about compound downselection using their own cost and utility functions," Fromer says.
In the future, the researchers want to incorporate additional complexity into SPARROW. For instance, they'd like to enable the algorithm to consider that the value of testing one compound may not always be constant. They also want to include more elements of parallel chemistry in its cost-versus-value function.
"The work by Fromer and Coley better aligns algorithmic decision making to the practical realities of chemical synthesis. When existing computational design algorithms are used, the work of determining how to best synthesize the set of designs is left to the medicinal chemist, resulting in less optimal choices and extra work for the medicinal chemist," says Patrick Riley, senior vice president of artificial intelligence at Relay Therapeutics, who was not involved with this research.
"This paper shows a principled path to include consideration of joint synthesis, which I expect to result in higher quality and more accepted algorithmic designs."
"Identifying which compounds to synthesize in a way that carefully balances time, cost, and the potential for making progress toward goals while providing useful new information is one of the most challenging tasks for drug discovery teams.
"The SPARROW approach from Fromer and Coley does this in an effective and automated way, providing a useful tool for human medicinal chemistry teams and taking important steps toward fully autonomous approaches to drug discovery," adds John Chodera, a computational chemist at Memorial Sloan Kettering Cancer Center, who was not involved with this work.
More information: Jenna C. Fromer et al, An algorithmic framework for synthetic cost-aware decision making in molecular design, Nature Computational Science (2024). DOI: 10.1038/s43588-024-00639-y
Journal information: Nature Computational Science
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.