Genome editing, a next step in genetic therapy, corrects hemophilia in animals
Using an innovative gene therapy technique called genome editing that hones in on the precise location of mutated DNA, scientists have treated the blood clotting disorder hemophilia in mice. This is the first time that genome editing, which precisely targets and repairs a genetic defect, has been done in a living animal and achieved clinically meaningful results.
As such, it represents an important step forward in the decades-long scientific progression of gene therapy -- developing treatments by correcting a disease-causing DNA sequence. In this new study, researchers used two versions of a genetically engineered virus (adeno-associated virus, or AAV) -- one carrying enzymes that cut DNA in an exact spot and one carrying a replacement gene to be copied into the DNA sequence. All of this occurred in the liver cells of living mice.
"Our research raises the possibility that genome editing can correct a genetic defect at a clinically meaningful level after in vivo delivery of the zinc finger nucleases," said the study leader, Katherine A. High, M.D., a hematologist and gene therapy expert at The Children's Hospital of Philadelphia. High, a Howard Hughes Medical Institute Investigator, directs the Center for Cellular and Molecular Therapeutics at Children's Hospital, and has investigated gene therapy for hemophilia for more than a decade.
The study appeared online today in Nature.
High's research, a collaboration with scientists at Sangamo BioSciences, Inc., makes use of genetically engineered enzymes called zinc finger nucleases (ZFNs) that act as molecular word processors, editing mutated sequences of DNA. Scientists have learned how to design ZFNs custom-matched to a specific gene location. ZFNs specific for the factor 9 gene (F9) were designed and used in conjunction with a DNA sequence that restored normal gene function lost in hemophilia.
By precisely targeting a specific site along a chromosome, ZFNs have an advantage over conventional gene therapy techniques that may randomly deliver a replacement gene into an unfavorable location, bypassing normal biological regulatory components controlling the gene. This imprecise targeting carries a risk of "insertional mutagenesis," in which the corrective gene causes an unexpected alteration, such as triggering leukemia.
In hemophilia, an inherited single-gene mutation impairs a patient's ability to produce a blood-clotting protein, leading to spontaneous, sometimes life-threatening bleeding episodes. The two major forms of the disease, which occurs almost solely in males, are hemophilia A and hemophilia B, caused respectively by a lack of clotting factor VIII and clotting factor IX. Patients are treated with frequent infusions of clotting proteins, which are expensive and sometimes stimulate the body to produce antibodies that negate the benefits of treatment.
In the current study, the researchers used genetic engineering to produce mice with hemophilia B, modeling the disease in people. Before treatment, the mice had no detectable levels of clotting factor IX.
Previous studies by other researchers had shown that ZFNs could accomplish genome editing in cultured stem cells that were then injected into mice to treat sickle cell disease. However, this ex vivo approach is not feasible for many human genetic diseases, which affect whole organ systems. Therefore the current study tested whether genome editing was effective when directly performed in vivo (in a living animal).
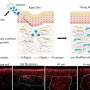
High and colleagues designed two versions of a vector, or gene delivery vehicle, using adeno-associated virus (AAV). One AAV vector carried ZFNs to perform the editing, the other delivered a correctly functioning version of the F9 gene. Because different mutations in the same gene may cause hemophilia, the process replaced seven different coding sequences, covering 95 percent of the disease-carrying mutations in hemophilia B.
The researchers injected mice with the gene therapy vector, which was designed to travel to the liver—where clotting factors are produced. The mice that received the ZFN/gene combination then produced enough clotting factor to reduce blood clotting times to nearly normal levels. Control mice receiving vectors lacking the ZFNs or the F9 minigene had no significant improvements in circulating factor or in clotting times.
The improvements persisted over the eight months of the study, and showed no toxic effects on growth, weight gain or liver function, clues that the treatment was well-tolerated.
"We established a proof of concept that we can perform genome editing in vivo, to produce stable and clinically meaningful results," said High. "We need to perform further studies to translate this finding into safe, effective treatments for hemophilia and other single-gene diseases in humans, but this is a promising strategy for gene therapy." She continued, "The clinical translation of genetic therapies from mouse models to humans has been a lengthy process, nearly two decades, but we are now seeing positive results in a range of diseases from inherited retinal disorders to hemophilia. In vivo genome editing will require time to mature as a therapeutic, but it represents the next goal in the development of genetic therapies."
More information: "In vivo genome editing restores hemostasis in a mouse model of hemophilia," Nature, published online June 26, 2011. doi: 10.1038/nature10177
Provided by Children's Hospital of Philadelphia