New method takes snapshots of proteins as they fold
 and the beta sheet (the flat ribbon) in the image to the right. These secondary structures then double back on themselves to form the final structure.")
(PhysOrg.com) -- People have only 20,000 to 30,000 genes (the number is hotly contested), but they use those genes to make more than 2 million proteins. It's the protein molecules that domost of the work in the human cell. After all, the word protein comes from the Greek prota, meaning "of primary importance."
Proteins are created as chains of amino acids, and these chains of usually fold spontaneously into what is called their "native form" in milliseconds or a few seconds.
A protein's function depends sensitively on its shape. For example, enzymes and the molecules they alter are often described as fitting together like a lock and key. By the same token, misfolded proteins are behind some of the most dreaded neurodegenerative diseases, such as Alzheimer's, Parkinson's and mad cow disease.
Scientists can't match the speed with which proteins fold. Predicting how chains of amino acids will fold from scratch requires either powerful supercomputers or cloud sourcing (harnessing the pattern recognition power of thousands of people by means of games such as Folding@home).
Either way, prediction is time-consuming and often inaccurate, so much so that the protein-structure bottleneck is slowing the exploitation of DNA sequence data in medicine and biotechnology.
A clever way of watching proteins fold and unfold may finally provide the kind of detail needed to improve protein structure predictions.
In a recent issue of the online version of the Journal of the American Chemical Society three scientists, led by Michael L. Gross, PhD, professor of chemistry in Arts & Sciences and of medicine and immunology in the School of Medicine at Washington University in St. Louis, describe a proof-of-principle study in which they use the new approach to watch the folding of a small protein called barstar.

What they do is roughly analogous to filming flying bullets or bursing balloons with a stroboscope and a fast camera. The "stills" taken by the camera slow motion to the point that normally imperceptible events are laid open to scrutiny.
The scientists are using a version of this old trick to "watch" proteins fold. The "strobe light" is a temperature jump and the "camera" is a fast chemical reaction whose outcome is measured by a sensitive mass spectrometer.
Why folding is a complex problem
One of the dogmas of modern biology is that the sequence of amino acids determines how a protein will fold. If the amino acid sequence is known, it should be possible to calculate the protein's final structure from scratch.
But like many things in life, it's harder than it looks.
"Think of a protein as thousands of atoms connected together by springs," says Gross, who is also director of the National Institutes of Health/ National Center for Research Resources (NIH/NCRR) Mass Spectrometry Resource "If you were to suspend this object with a string from the ceiling and let it flop around, imagine how many shapes it could take."
"An enormous number, because it is free to move in so many different ways."
In practice, scientists often predict protein structure not from scratch but by analogy. They sift through large databases for proteins with similar sequences of amino acids and assume similar amino-acid chains will fold in similar ways.
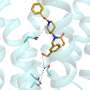
 is to bind tightly to barnase (left), preventing it from chopping up RNA, a molecule necessary to life, until it is secreted from the cell.")
"But," says Gross, "at some point any method for predicting protein structure has to be checked against experimental evidence that shows how proteins actually do fold."
That's what his research is all about.
A model protein for the experiment
Barstar is a small protein synthesized by a soil bacterium that is often used in folding studies.
Importantly, barstar's "native state" is known, as is its primary structure, the sequence of the protein subunits called amino acids of which it is made. What isn't known is how the amino-acid chain twists and coils to form the final structure.
Fortunately for the scientists, barstar, unlike most proteins, is unfolded at zero degrees Celsius and begins to fold as it warms.
The folding takes place in microseconds (thousandths of a second).
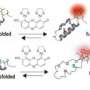
, and hydrogen peroxide (paired red dots) is injected into a capillary. The solution is hit with a laser at an infrared wavelength (second image), which heats the protein enough to encourage it to start folding (the blue coil is an alpha helix, a common protein \"fold\"). In the third step, the solution is exposed to an ultraviolet pulse that breaks the hydrogen peroxide into two parts that attach oxygen atoms (red dots) to portions of the protein that are still outside the folding structure and exposed to the solution. The protein is later cut up and the parts \"weighed\" to see if they are carrying additional oxygen atoms. The pathway by which the protein folds can be deduced by repeatedly \"painting\" it with oxygen in this way. Credit: Nature Publications")
How the method works
The scientists begin by injecting a cold solution of barstar and a tiny amount of hydrogen peroxide into an optical fiber so thin it is difficult to believe it is actually hollow.
"Plugs" of sample in the fiber are then hit with two laser pulses in quick succession.
The first pulse, called a T jump, heats the solution just enough to make a different protein conformation energetically favorable.
The second pulse then breaks some of the hydrogen peroxide (H2O2) molecules into two haves, each of which is a very reactive hydroxyl (-OH) radical.
The radicals react with those parts of the protein that are exposed to the solution, "painting" them with oxygen atoms.
"Imagine," says Gross, "that you suspended a styrofoam model of a partially folded protein and spray-painted it blue. The outside parts would be painted blue; those buried within would remain white."
The radical reactions must be terminated rapidly; otherwise some "painting" may occur within the structure. Within a microsecond, a scavenger amino acid clears away any remaining hydroxyl radicals to prevent them from breaking bonds and altering the protein's configuration.
The same process is repeated 500 times, taking rapid-fire "snapshots" of the protein's quickly changing confirmation.
"The hydroxyl radicals don't mark everything," says Gross. "But they mark about half the amino acids, which is really pretty good. Most other chemical reagents are too specific and too slow for this experiment. Compared to hydroxyl radicals they're just plain ponderous."
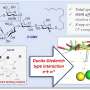
Weighing the painted proteins
"We collect each drop of marked protein as it emerges from the fiber," says Gross. "Then we digest the protein very slowly and carefully with an enzyme that cleaves the amino acid chains at specific locations, creating a known set of protein fragments, called peptides
These protein fragments are separated according to type by liquid chromatography, and a mass spectrometer then "weighs" each fragment type to see whether it has picked up oxygen atoms.
"Detecting an extra oxygen is child's play for a modern mass spectrometer," says Gross. "Most instruments can even detect an extra proton, with is one-sixteenth the mass of an oxygen atom."
"In the same instrument, on the fly, we break apart the protein fragments and again 'weigh' the bits to see which one still carries the oxygen atom. This lets us deduce the oxygen's location on the original fragment."
By following barstar to its first intermediate state, or way station enroute to its native state, the scientists demonstrated that the new technique can follow folding and unfolding on a submillisecond time scale.
'Massive amounts of detail'
Gross is the first to say that this proof-of-principle experiment stands at the end of a long line of elegant experiments of a similar type, called pump-probe experiments.
Other techniques probe the change in protein structure by monitoring the absorption or emission of light--or a similar physical effect. They can provide only global information, such as the rate constant of a folding reaction.
"Because we use a chemical rather than a physical probe, we can see what's going on in much greater detail," says Gross. "We can say which part of the structure closes first, which second, and so on."
The new technique caught the attention of protein scientist Martin Gruebele of the University of Illinois, who spotlighted it in the Dec. 2, 2010, issue of the journal Nature.
It "could provide truly massive amounts of detail about fast protein folding," wrote Gruebele, which might finally allow scientists "to correctly predict the biologically active structure of a protein starting from the unfolded state."
Provided by Washington University in St. Louis