Same disease, different stem cell models
In the last three years, a new technique for reprogramming adult cells has given scientists an easier and less controversial way to harness the power of embryonic-like stem cells to study human disease from its earliest beginnings in hopes of gleaning new insights into the root causes of disease and developing new therapies.
But the reprogrammed cells, known as induced pluripotent stem (iPS) cells, are different from embryonic stem cells in their ability to model a human genetic disease, a new cell-to-cell comparison shows.
"This is the first example where we can clearly show induced pluripotent stem cells and embryonic stem cells behave differently in a disease model," said co-senior author George Daley, Director of the Stem Cell Transplantation Program at Children's Hospital Boston. Daley's team has turned patient cells back into stem cells for a range of diseases.
In the new study in the May 7 Cell Stem Cell, the researchers made iPS cells from the skin cells of three patients with fragile X syndrome, the most common form of inherited mental retardation in boys. By almost every measure, virtually the entire genome was dialed back in time. The key exception was the disease-causing gene, which becomes inactivated to cause the disease, and did not get turned back on in the iPS cells.
"Both iPS and embryonic stem cell lines have the same mutation," said co-senior author Nissim Benvenisty, director of the Stem Cell Unit at the Hebrew University of Jerusalem, whose lab established an embryonic stem cell model of fragile X syndrome three years ago at a time when such research was severely restricted in the United States. "However, we saw a difference between the two systems."
About one-third of children with fragile X have behavioral symptoms that overlap with autism. Scientists hope a stem cell model of fragile X will help them study what goes wrong and test drugs that may help treat both abnormal conditions.
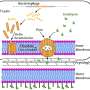
"It's known that the fragile X protein regulates the expression of receptors at the synapse between nerve cells," Daley said. "In the absence of the protein, nerve cells express too much of an excitatory receptor."
The mutation that silences the fragile X gene and blocks its protein lies buried just upstream of the gene's coding region in triplet repeats of DNA. Normally, that region harbors up to 50 CGG repeats. Fragile X syndrome occurs when the repeats number more than 200.
The mutation alone is not enough to cause disease. "In rare cases, people can have the full mutation, but the gene is still expressed," said co-first author Achia Urbach, a postdoctoral fellow in the Daley lab at Children's and former graduate student in the Benvenisty lab in Jerusalem.
Three years ago, Urbach, Benvenisty and their Israeli collaborators reported the first direct evidence that the fragile X gene was silenced upon differentiation. The fragile X gene, called FMR1, remains active until cells begin to differentiate in preparation for forming different tissues and organs. Somehow, that process locks down the DNA with epigenetic changes that prevent the gene from being transcribed. To study the FMR1 gene silencing in embryonic stem cells, they established embryos affected by the fragile X condition in collaboration with Lis Maternity Hospital in Tel Aviv. The affected embryos were identified by preimplantation diagnosis, resulting from IVF treatment of a woman who carried the fragile X mutation and wanted healthy babies, Urbach said.
After that paper, Urbach joined the Daley lab to study iPS cells. Beginning with patients' cells, Daley's group has developed more than a dozen disease-specific stem cell lines. "We thought it would be interesting to compare the two systems," said Urbach of the embryonic and induced fragile X stem cells.
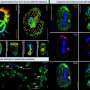
Urbach and co-first author Ori Bar-Nur, a graduate student in Benvenisty's lab first created fragile X iPS cells from the skin cells of two affected patients and related lung cells from a 22-week-old fetus with fragile X, testing them extensively to assure themselves and other researchers of their stem-cell qualities. They also investigated why the FMR1 gene remained stubbornly locked down.
"We show the reason the gene is not expressed is because it still has the epigenetic markers for silencing," Urbach said.
The fragile X gene may be one of the first genes to resist the reprogramming process that transforms adult tissue cells into iPS cells, but it's likely not the last, said the researchers.
"This raises a general caution for using iPS as a faithful reflection of a disease process," Daley said. "There are lots of conditions where you have gene defects that lead to gene silencing. Such conditions may not be faithfully modeled by iPS cells. Fragile X is a disease where using embryonic stem cells as a tool is essential."
The differences in the iPS and embryonic fragile X stem cells make them useful for different types of studies, the researchers say. "On one hand, iPS cells are not as good for modeling the inactivation of the gene," Benvenisty said. "On the other hand, they may be a better model for studying neurons lacking expression of the gene."
"New insights into fragile X have stimulated clinical trials of drugs that block the overactive excitatory receptors in nerve cells," Daley said. "Early results hint that these drugs might ameliorate the condition of fragile X. With our stem cell models—diseases in a dish, if you will—we can test whether the drugs will reverse abnormal connections at the synapses that we think are at the basis of this condition. If your goal is blocking FMR1 gene silencing, you're better off working on drug screens in embryonic stem cells than in iPS cells."
Provided by Children's Hospital Boston