Researchers study signaling networks that set up genetic code
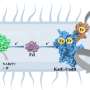
 maintain the genetic code by charging tRNA (brown) with the cognate amino acid. Upon tRNA recognition, allosteric signal is passed between the anticodon and the distant catalytic site (colored spheres). The paths for signal transduction (green) in a network of dynamical contacts are weighted by their frequency of occurrence.")
(PhysOrg.com) -- In a new study, researchers at the University of Illinois have identified and visualized the signaling pathways in protein-RNA complexes that help set the genetic code in all organisms. The genetic code allows information stored in DNA to be translated into proteins.
The researchers report their findings in a paper accepted for publication in the Proceedings of the National Academy of Sciences, and posted on the journal's Web site.
"Using molecular dynamics simulations and new visualization software, we can examine and compare different signaling pathways found in living organisms," said Zaida Luthey-Schulten, a William and Janet Lycan Professor of Chemistry at the U. of I., and the paper's corresponding author. "Our methodology is applicable to all protein-RNA complexes involved in translation."
Translation is an essential process in living cells whereby messenger RNA (mRNA) serve as templates in the ribosome to produce specific proteins. How a signal moves through the ribosome during translation has not been understood.
In the first step of translation, an amino acid must be added onto the end of the correct transfer RNA (tRNA) containing the corresponding anticodon according to the genetic code. (The anticodon is a sequence of three nucleotides that can bind to a specific sequence of three nucleotides - the codon - in mRNA.)
The proteins that attach the correct amino acid to the tRNA are the aminoacyl-tRNA synthetases. There are 20 different aminoacyl-tRNA synthetases in an organism typically, one for each kind of amino acid.
From a pool of thousands of amino acids and tRNAs in the cell, each aminoacyl-tRNA synthetase must recognize and pair the correct amino acid and tRNA. After this pairing has occurred, the tRNA is transported to the ribosome where the messenger RNA obtained from the DNA sequence is translated into a protein.
How the signal is transmitted from the anticodon on the tRNA at one end of the protein-tRNA complex to the site of chemical reaction in the aminoacyl-tRNA synthetase at the other end has puzzled scientists for a long time. Now, using network analysis algorithms in combination with molecular dynamics simulations, the U. of I. researchers have been able to identify the network of interactions.
The network algorithms, developed originally by computer scientists to study the communication patterns in the World Wide Web, air travel, and the spread of disease, were adapted to find the optimal and suboptimal communication pathways.
"The signaling pathways in the protein-tRNA complexes are analogous to air travel, where passengers departing from small airports pass through major hubs on their way to distant destinations," Luthey-Schulten said. "So too in biology, modules of amino acids and nucleotides communicate using paths that pass through a few important interconnecting links."
To study those links using network analysis algorithms, the researchers represented each amino acid and nucleotide in the protein-RNA complex as a node. Edges connected pairs of nodes, and the network consisted of nodes with connecting edges.
Variations in the connectivity of the network give rise to modules, which can be viewed as local communities in the network, the researches report. Nodes within the same community are highly interconnected and can communicate through a large number of paths with a small difference in distance.
To further analyze the protein-tRNA network, the researchers "weighted" the edges by correlation values derived from molecular dynamics simulations.
"The optimal path is the shortest path, the most correlated path along which the information is transferred," said graduate student and co-author John Eargle. "There are other paths nearby that are also highly correlated, so the optimal path is not the only path where communication is being taken through the protein."
Visualization of the communication pathways allowed the researchers to study the redundancy of the pathways and the effects of mutations on the signaling pathways that set the genetic code.
"With our visualization software, we also can overlap the optimal paths from different proteins and compare networks," Luthey-Schulten said. "The ability to rapidly visualize and compute the networks will accelerate the design efforts to expand the genetic code to include novel amino acids and allow researchers to apply the technique to much larger systems like the ribosome."
The enhanced visualization software is part of VMD (Visual Molecular Dynamics), a program for visualizing and analyzing molecular dynamics simulations. Developed at the U. of I. and distributed free of charge, VMD is designed to efficiently handle large three-dimensional systems containing more than a million atoms.
Source: University of Illinois at Urbana-Champaign (news : web)