Sometimes the average just isn't good enough

Computational biologists from the Max F. Perutz Laboratories of the University of Vienna and the Medical University of Vienna show that averaging is not always a good thing when it comes to analyzing protein crystal structures. The recent publication by Bojan Zagrovic and his team in Nature Communications shows that protein structures could be more dynamic and heterogeneous than current methods of X-ray analysis suggest.
When averaging is good and when it's not
Usually averaging is a good thing that can make life a lot easier. For example, when you eat out with a group of friends and it comes to paying. If everyone had a meal and a drink and you split the bill total by the number of people, everyone will pay pretty close to what they would have paid for their individual meal and drink. However, if some people had a starter, a steak for main, a dessert and champagne while you had spaghetti and a soft drink, you will feel pretty much ripped off when you pay the average of 45€ for your meal. In science, averaging is a good thing too – researchers often repeat their experiment several times and average the results. As long as the results are within a certain level of deviation, the scientists can then be confident that what they found in the lab is reliable.
Determining protein structures: X-ray crystallography
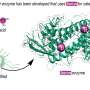
One of the most important methods in biology is X-ray crystallography, which is used to analyze protein structures. Knowing such structures allows scientists to draw conclusions about what a protein does and how it does it, but also to develop medicines that inhibit or activate that protein's function. For X-ray crystallography, proteins are purified and dried to form a crystal, each of which contains millions of copies of the same protein. Shining X-rays on the crystal then allows conclusions about where the smallest building blocks of a protein – the atoms – are located and how dynamic each of them is, i.e. how much it can wiggle around in its location. By doing that for one crystal they get averages, which are based on the behavior of millions of copies of equivalent atoms. You would think this is enough to be confident that this is what the protein looks like in nature too. But it's actually not and sometimes averaging can be misleading as Bojan Zagrovic, lead author of the study, explains: "Take, for example, the average location of a goalie during a football match. Considering that the teams switch sides at halftime, it is roughly at the center of the field, a clearly non-representative situation."
Atoms in proteins wiggle up to six fold more than currently thought
So how correctly do current programs for the analysis of X-ray crystallographic data capture a protein's structure and its dynamics? This was one of the questions Antonija Kuzmanic wanted to answer during her PhD studies with Zagrovic and supported by his European Research Council (ERC) Starting Grant. Together with collaborator Navraj S. Pannu of Leiden University, The Netherlands, she used computer simulation to "built" a protein crystal and analyzed it by the methods of X-ray crystallography, before using standard software programs to capture the protein's features from crystallographic data. This allowed her to test if the way crystallographic data is currently analyzed "sees" what's really there. "We were really surprised to find that current software programs, used to predict a protein's structure from X-ray crystallography data, underestimates the level of dynamics – so how much each atom can wiggle around in its position – by up to six fold. This is a lot, it's like if we could suddenly turn our head 180 degrees rather than just to the left or right", Antonija Kuzmanic explains.
Inspirational work
Garib Murshudov of the University of Cambridge, UK, structural biologist and one of the examiners of Antonija Kuzmanic's PhD thesis, wrote: "This is my favorite chapter, it is inspirational … it clearly shows that it is necessary to design new ways to describe protein dynamics in crystals." More accurate ways to interpret X-ray crystallography data and determine the dynamics of a protein will not only give a clearer and a more realistic picture of what the protein looks like in nature – so that goalie's location and motions are precisely known – but will also help to develop medicines that can modify the protein's function more accurately and more potently.
More information: Antonija Kuzmanic, Navraj S. Pannu and Bojan Zagrovic: X-ray refinement significantly underestimates the level of microscopic heterogeneity in biomolecular crystals. In: Nature Communications (January 2014). DOI: dx.doi.org/10.1038/ncomms4220
Journal information: Nature Communications
Provided by University of Vienna