Lessening X-ray damage is healthy for protein discovery data too

(Phys.org) —New recommendations for using X-rays promise to speed investigations aimed at understanding the structure and function of biologically important proteins – information critical to the development of new drugs. Scientists from two U.S. Department of Energy national laboratories, Argonne and Brookhaven, and the University of Washington, Seattle, evaluated options to remedy problems affecting data collection in their new study.
Scientists who use powerful X-ray beams to study protein crystals face a dilemma: the beams provide the best tool for understanding a protein's structure and biological function, but they often damage the crystal, which may require repeated experiments that add time and cost to the research.
"Although X-ray crystallography is the go-to technique for determining protein structure and function, it is not without problems," said Andrzej Joachimiak at Argonne. The use of powerful X-ray beams causes radiation damage resulting in loss of data and the weak diffraction of crystals. In the end, this leads to an incomplete picture of the structure, and of how molecules interact with each other and their environment.
"The problem occurs when a protein crystal absorbs energy from incoming X-rays, which emits electrons that destroy or alter parts of the sample," Joachimiak said.
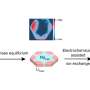
The research team examined three different X-ray-based methods for solving protein structures and recommended one called "submicrometer line focusing" as the most promising for easing the dilemma. As its name suggests, the beam strikes the protein crystal with an area smaller than a micrometer, or smaller than one thousandth of a millimeter. The tiny impact area minimizes damage. Also like its name, the beam is focused as a vertical line, delivering a more concentrated dose of X-rays per area.
The researchers also suggested using a new lens they designed that breaks the powerful beam into many mini-beams, spaced far enough apart that the damage one mini-beam creates lies outside the area probed by neighboring mini-beams.
"By carefully spacing the beams, we can lessen the damage, gather better data, and do it faster," said Joachimiak. "And because there are several beams rather than just one being released simultaneously, we can collect a greater quantity of useful data."
Synchrotron facilities like the DOE's Advanced Photon Source (APS) at Argonne contain particle accelerator systems designed to produce extraordinarily bright and high-energy X-ray beams. At these facilities, scientists can peer deeply into the atomic structure of molecules using the method of X-ray crystallography.
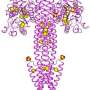
Scientists need to see what molecules, particularly proteins, look like. While molecules are too small to be visualized directly, their shape can be reconstructed by looking at the patterns of how X-rays diffract, or scatter, off them. Crystallography uses crystals of proteins because crystals have repeating patterns that give scientists enough data to reconstruct the exact shape as a 3-D model. From this form, scientists can often determine chemical interactions and processes that can be used to design pharmaceuticals.
In this study, the team looked at the penetration depth of the damaging electrons and at the spread or distribution of the damaged area on the protein crystal. While earlier studies recognized the issue, this team is the first to collect high-resolution data and directly measure the damaged area using a line focus beam. The team also found that earlier work had underestimated the depth of the problem.
The findings will aid synchrotron researchers as they continue to develop more brilliant and powerful instruments such as those at the APS.
The results and recommendations are described in a paper published in August in Acta Crystallographica Section D: Biological Crystallography, titled "Mitigation of X-ray damage in macromolecular crystallography by submicrometre line focusing."
More information: "Mitigation of X-ray damage in macromolecular crystallography by submicrometre line focusing." Finfrock YZ, Stern EA, Alkire RW, Kas JJ, Evans-Lutterodt K, Stein A, Duke N, Lazarski K, Joachimiak A. Acta Crystallogr D Biol Crystallogr. 2013 Aug. DOI: 10.1107/S0907444913009335.
Provided by Argonne National Laboratory