An unlikely route to ferroelectricity
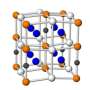
”] with polar bilayers previously assumed for LuFe2O4. Right side: The refined LuFe2O4 monoclinic crystal structure containing charged bilayers. For both configurations, the Fe atoms are arranged in Fe/O bilayers separated by Lu single layers. The different Fe valances are indicated by different colors. The brackets beside (each) indicate the majority charge configuration for each single layer. Here, for the polar model (left side), from the charge imbalance a net electric polarization is present; in contrast the model on the right side contains non-polar bilayers. Furthermore, the red arrows indicate the spin structure.")
(Phys.org) -- Ferroelectricity, which was first observed in the 1940s, is an interesting phenomenon involving the spontaneous (non-induced) formation of charge polarization (separation of charge) in certain materials. This is analogous to the spontaneous formation of magnetic fields in iron and other elements via ferromagnetism. Multiferroics (materials exhibiting both ferroelectricity and ferromagnetism) have attracted increased interest of late due to their potential use in various technologies, such as improved electronic memory chips and highly sensitive magnetic field sensors. The crystalline material lutetium-iron-oxide (LuFe2O4) has, in turn, garnered much attention due to its purported multiferroic properties.
A team of researchers from the Jülich Centre for Neutron Science and Argonne National Laboratory utilized several techniques, including studies at the U.S. Department of Energy Office of Science’s Advanced Photon Source (APS) at Argonne, and bond-valance-sum analysis and the charge-spin coupling of valence electrons to unmask the actual charge ordering within LuFe2O4 to determine the exact source of its purported ferroelectric behavior.
Previous laboratory measurements and theoretical considerations had indicated the most likely charge ordering (the microscopic localization of electric charges) within LuFe2O4 was due to polarized iron/oxygen (Fe/O) bilayers.
Using an “assumption-free” approach, the research team’s application of multiple techniques unmasked the actual charge ordering within LuFe2O4. Against all expectations, charge ordering does not arise from polar bilayers, but rather is due to charge separation on neighboring Fe/O bilayers. This finding means that the reported ferroelectric behavior of LuFe2O4 is highly unlikely.
Crystalline LuFe2O4 consists of alternating stacks of one lutetium oxide (Lu/O) and two ferrous oxide (Fe/O) planes or layers (see left side of the figure). According to the conventional model depicted in the left half of the figure, charge ordering in Fe/O bilayers (a pair of adjacent Fe/O planes) leads to an electric dipole moment perpendicular to the Fe/O planes.
The iron-oxygen bonds within the bilayer are predominately ionic, featuring iron atoms in the Fe+2 and Fe+3 valence states. The superscripts refer to the number of electrons “donated” by individual iron atoms to the Fe/O molecular bonding.
Note that the top Fe/O layer in the figure (left side) has more Fe+2 than Fe+3 valence states, while the reverse is true for the bottom Fe/O layer. The different valence predominance in the two ferrous oxide layers (one rich in Fe+2, with the other rich in Fe+3) produces the polarization (separation) of charges giving rise to an electric field. This pattern repeats multiple times in the crystal, producing large-scale (macroscopic) ferroelectricity.
This conventional model, however, was never proven directly by any previous research.
To pin down the exact distribution of iron valence states within the Fe/O bilayers, and settle the question of whether or not they are polar, the researchers used bond-valance-sum analysis, which was linked to x-ray diffraction data of single LuFe2O4 crystals.
The Argonne X-ray Science Division (XSD) 6-ID-D high-energy station at the APS at Argonne was used for the x-ray diffraction study. The picture that emerged showed two possible, but diametrically opposed charge-ordering structures: one featuring polarity within the Fe/O bilayers, the other with non-polar Fe/O bilayers featuring net charges on distinct bilayers.
The structure of Fe valence states within the Fe/O bilayers was further refined by the use of x-ray magnetic circular dichroism (XMCD) carried out at the XSD 4-ID-C beamline at the APS. XMCD uses right- and left-circularly polarized x-rays to determine electron spin states in a material immersed in a magnetic field. X-ray absorption is carried out at the iron atoms' L-edge, which is the energy value at which electrons residing in the L orbital of the Fe atoms are just capable of absorbing photons.
LuFe2O4 crystals were subjected to a 4-T magnetic field. The XMCD data revealed that all spins of the Fe+2 ions were oriented in the direction of the magnetic field, as were one-third of the Fe+3 spins. Two-thirds of the Fe+3 spins were aligned opposite to the magnetic field.
Combining this result with the spin-structure determined earlier, only 28 possible charge configurations were possible, with only one consistent with x-ray diffraction. The result is the charge ordering structure depicted in the right side of the figure.
Conventional wisdom has held that LuFe2O4 provided a clear (and the only known) example of an oxide compound exhibiting ferroelectricity via charge ordering. However, the detailed picture of charge ordering revealed by this research is incompatible with a polar structure within the Fe/O bilayers. Instead, a more complex charge ordering structure has emerged, one that is non-polar and unaffected by external electric fields, and hence incompatible with any ferroelectric behavior in LuFe2O4.
The apparent lack of multiferrocity in LuFe2O4 undoubtedly comes as a disappointment to those looking to exploit the potential of such materials. According to article co-author Manuel Angst, who initiated this research, “Charge-order based multiferroicity is very attractive from the point of view of applications, but as these results show, an actual example has yet to be found.”
Nevertheless, the unexpected structure elucidated for this rare-earth iron oxide is interesting in its own right. For one thing, the way in which bilayers rich in Fe+2 and Fe+3 produce charge ordering is quite unexpected, suggesting that similar mechanisms for charge ordering in other crystalline materials are awaiting discovery. Additionally, theorists must now attempt to answer how charge transfer occurs between the Fe/O bilayers of this material, across a distance of some 6 Å (0.6 nm).
More information: J. de Groot, et al. “Charge order in LuFe2O4: an unlikely route to ferroelectricity,” Phys. Rev. Lett.108, 187601 (2012). DOI: 10.1103/PhysRevLett.108.187601
See also:
M. Angst, et al., Phys. Rev. Lett. 101, 227601 (2008).
X.S. Xu, et al., Phys. Rev. Lett. 101, 227602 (2008).
J. de Groot, et al., Phys. Rev. Lett. 108, 037206 (2012).
Journal information: Physical Review Letters
Provided by Argonne National Laboratory