This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Calculating faster: Coupling AI with fundamental physics
. DOI: 10.1038/s41467-024-50620-6")
Atoms are complex quantum systems consisting of a positively charged nucleus surrounded by negatively charged electrons. When multiple atoms come together to form a molecule, the electrons of the constituent atoms interact in a complicated manner, making the computer simulation of molecules one of the hardest problems in modern science.
Researchers from the Berlin Institute for the Foundations of Learning and Data (BIFOLD) at TU Berlin and Google DeepMind have now developed a novel machine learning algorithm which enables highly accurate simulations of the dynamics of a single or multiple molecule on long time-scales. Their work has now been published in Nature Communications.
These so-called molecular dynamics simulations are important to understand the properties of molecules and materials and have potential applications in drug development and material design (e.g. for use in solar panels and batteries). Traditional methods to compute the interactions of electrons rely on finding solutions of the so-called Schrödinger equation.
The Schrödinger equation describes the energy levels that a quantum system—e.g. atoms or molecules—can assume. This is a notoriously difficult task, and finding a solution for molecules containing more than a few dozen atoms may take several days—even on powerful computers. To make matters worse, for running molecular dynamics simulations over long time-scales, the Schrödinger equation needs to be solved thousands or even millions of times, making the computational cost quickly exceed the compute resources that are available today.
"The simulation of such interactions and the resulting predictions for complex processes like protein folding or the binding between individual molecules is a long-held dream of many chemists and material scientists, and would save many expensive and labor-intensive experiments," explains BIFOLD researcher Thorben Frank.
In recent years, machine learning (ML) methods have brought this dream within reach. Instead of explicitly solving the Schrödinger equation, they can learn to directly predict the overall outcome of the relevant electronic interactions at the atomistic level, with greatly reduced computational cost.
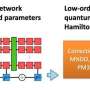
The difficulty is then shifted to finding efficient algorithms for "teaching" the machine learning system how the electrons interact without modeling them explicitly. To reduce the complexity of this task, many learning algorithms use the fact that physical systems follow so-called invariances.
Simply put, certain properties of molecules stay the same when molecules are moved in space but the relative distances between individual atoms stay fixed—meaning the machine does not need to learn anything new in these cases. However, the way these invariances are typically incorporated into ML models is computationally expensive, ultimately limiting the speed with which the models can perform molecular dynamics simulations.
To address this shortcoming, the BIFOLD scientists have devised a new learning algorithm that decouples invariances from other information about a chemical system at the outset. Unlike previous methods that required extracting invariant components from each operation within the model, this new approach simplifies the process. Now, the ML model can reserve the most complex operations for the physical information that really matters and drastically reduce the overall computational cost.
"Simulations that required months or even years of computation on high-performance computer clusters, can now be performed within a few days on a single computer node. The leap in efficiency allows long-time scale simulations, which are necessary for understanding the structure, dynamics and functioning of atomistic systems. It thus enables deeper insights into the most complex and fundamental processes of nature," says BIFOLD researcher Dr. Stefan Chmiela who spearheaded the research project.
In the future, the accurate simulation of the interaction of molecules with proteins in the human body could allow researchers to develop new drugs without the need to perform experiments—saving time and money while at the same time being more environmentally friendly.
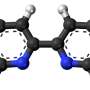
To showcase potential applications of the algorithm, the team used the new ML method to identify the most stable version of docosahexaenoic acid, a fatty acid which is a primary structural component in the human brain. This task requires scanning tens of thousands of potential candidates with high accuracy. So far, such an analysis would have been infeasible with traditional quantum mechanical methods.
As noted by Prof. Dr. Klaus-Robert Müller, BIFOLD co-director and Principal Scientist at Google DeepMind, "This work demonstrates the potential of combining advanced machine learning techniques with physical principles to overcome long standing challenges in computational chemistry. It continues a critical line of research which puts focus on scaling ML approaches towards realistic chemical systems of practical interest."
Dr. Oliver Unke, Senior Research scientist at Google DeepMind comments, "Earlier this year, we succeeded in scaling models to thousands of atoms, but with new advancements like this, moving to even larger numbers of atoms may become possible."
While simulations with tens to hundreds of thousands of atoms are now becoming accessible, some structures consist of millions of atoms or more. The next generation of algorithms will need to be able to simulate such system sizes accurately, which requires a correct description of additional, complex, long-range physical interactions.
More information: J. Thorben Frank et al, A Euclidean transformer for fast and stable machine learned force fields, Nature Communications (2024). DOI: 10.1038/s41467-024-50620-6
Journal information: Nature Communications
Provided by Technical University of Berlin