This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Scientist advance simulation of metal-organic frameworks with machine learning

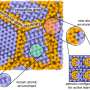
Hydrogen storage, heat conduction, gas storage, CO2 and water sequestration—metal-organic frameworks (MOFs) have extraordinary properties due to their unique structure in the form of microporous crystals, which have a very large surface area despite their small size. This makes them extremely interesting for research and practical applications. However, MOFs are very complex systems that have so far required a great deal of time and computing power to simulate accurately.
A team led by Egbert Zojer from the Institute of Solid State Physics at Graz University of Technology (TU Graz) has now significantly improved these simulations using machine learning, which greatly accelerates the development and application of novel MOFs. The researchers have published their method in npj Computational Materials.
Previously unrealistic to simulate with the accuracy of quantum mechanical methods
"To simulate certain properties of MOFs, it is necessary to simulate huge supercells. This applies, for example, to the calculation of heat conduction in MOFs, which is highly relevant for almost all applications," says Egbert Zojer, describing the challenge that had to be solved.
"The simulated supercells often contain tens of thousands or even hundreds of thousands of atoms. For these huge systems, it is then necessary to solve the equations of motion five to 10 million times. This is far beyond present day computational possibilities using reliable quantum mechanical methods."
Thus, until now, transferrable force fields often parametrized on the basis of experiments were often used for such calculations. However, the results obtained with such force fields turned out to be generally not sufficiently reliable.
This is now fundamentally changed by the use of machine-learned potentials. These are adapted to quantum mechanical simulations by utilizing a newly developed interplay of existing algorithms, including approaches developed at the University of Vienna. For the necessary material-specific machine learning of the potentials, the quantum mechanical simulations need to be carried out only for comparatively few and significantly smaller structures.
As a result, the calculations run many orders of magnitude faster and it is possible to simulate the forces in the huge supercells many millions of times on modern supercomputers. The decisive advantage here is that there is no relevant loss of accuracy compared to doing the simulations using quantum mechanical methods.
More efficient search for the desired properties
For the example of heat conduction of MOFs, this means that the newly developed simulation strategy will make it possible to simulate the relevant material properties even before the MOFs are synthesized, thus allowing researchers to reliably develop customized structures on the computer.
This represents a major leap forward for research into complex materials, which for heat transport will, for example, allow researchers to optimize the interaction between the metal oxide nodes and the semiconducting organic linkers. Using the new simulation strategy will also make it easier to overcome complex challenges. For example, MOFs must have good or poor thermal conductivity depending on their application.
A hydrogen storage system, for instance, must be able to dissipate heat well, while in thermoelectric applications good electrical conduction should be combined with the lowest possible heat dissipation.
In addition to simulating thermal conductivity, the new machine-learned potentials are also ideal for calculating other dynamic and structural properties of MOFs. These include crystallographic structures, elastic constants, as well as vibrational spectra and phonons, which play a decisive role in the thermal stability of MOFs and their charge transport properties.
"We now have tools that we know are incredibly efficient at providing us with reliable quantitative figures. This enables us to systematically change the structures of the MOFs in the simulations, while at the same time knowing that the simulated properties will be accurate. This will allow us, based on causality, to understand which changes in the atomistic structure generate the desired effects," says Egbert Zojer, who knows that research groups in Munich and Bayreuth have already taken up the new simulation strategy despite its recent publication.
More information: Sandro Wieser et al, Machine learned force-fields for an Ab-initio quality description of metal-organic frameworks, npj Computational Materials (2024). DOI: 10.1038/s41524-024-01205-w
Journal information: npj Computational Materials
Provided by Graz University of Technology