This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
Researchers reveal new method for calculating mechanical properties of solids using machine learning
. DOI: 10.1002/adts.202301171")
A research team from Skoltech introduced a new method that takes advantage of machine learning for studying the properties of polycrystals, composites, and multiphase systems. It attained high accuracy, nearly as good as that of quantum-mechanical methods, which are only applicable to materials with less than a few hundred atoms.
The new method also benefits from active learning on local atomic environments. The paper is published in the Advanced Theory and Simulations journal.
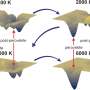
"Many industrial materials are synthesized as polycrystals or multiphase systems. They contain both a single crystal and amorphous components between single crystal grains. The large number of atoms makes it hard to calculate the properties of these systems using modern quantum-mechanical methods. Density functional theory can only be applied to materials with a few hundred atoms."
"To address the problem, we use a machine-learning approach based on Moment Tensor Potentials (MTP). These potentials have also been developed at Skoltech under the guidance of Professor Alexander Shapeev," commented Faridun Jalolov, the leading author of the study and a Skoltech Ph.D. student in the Materials Science and Engineering program.
As compared to other solutions, the authors see the potential of the new method in active learning on local atomic environments. When calculating a large structure with many hundreds of thousands of atoms, the MTP identifies which atom makes a mistake in the calculations, or is calculated incorrectly. The reason for this could be the limited training dataset, which prevents all possible system configurations from being considered.
A local environment of this atom is then "cut out," and its energy is calculated using quantum mechanics. Afterward, the data is added back to the training set for further learning. As the on-the-fly learning progresses, the calculations continue until they come across another configuration that needs to be included in the training process. Other known machine-learning potentials cannot be learned on small local parts of large structures, which limits their applicability and accuracy.
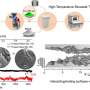
"As an example, we studied the mechanical properties of diamond polycrystals, which are the hardest naturally occurring materials and often used in industry—for example, when manufacturing drilling equipment for oil wells. The results show that the mechanical properties of these polycrystalline diamonds depend on the grain size—the bigger the grain, the more similar the properties are to those of a single crystal diamond," continued Jalolov.
The authors pointed out that this approach will allow for studying the mechanical properties of nonsingle crystalline materials that are typically synthesized and used in experiments, as well as conducting comprehensive studies of polycrystalline and composite materials and obtaining data as close to experimental results as possible.
"In actual use, materials that are not perfect crystals are frequently employed due to their inability for perfect crystals to meet the requirements of a specific piece of equipment fully."
"A good example of this is tungsten carbide and cobalt. By adding cobalt to tungsten carbide, the material becomes more crack-resistant, making it so valuable in applications. The new method will allow us to investigate the causes and ways of altering the mechanical properties of these multiphase systems on an atomic level," said Alexander Kvashnin, the head of the research and a professor at the Energy Transition Center.
More information: Faridun N. Jalolov et al, Mechanical Properties of Single and Polycrystalline Solids from Machine Learning, Advanced Theory and Simulations (2024). DOI: 10.1002/adts.202301171
Provided by Skolkovo Institute of Science and Technology