March 23, 2023 report
This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Predicting protein folding from single sequences with Meta AI ESM-2
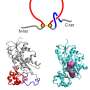
 Predicted contact probabilities (bottom right) and actual contact precision (top left) for PDB 3LYW. A contact is a positive prediction if it is within the top L most likely contacts for a sequence of length L. (B to D) Unsupervised contact prediction performance [long-range precision at L (P@L)] (SM A.2.1) for all scales of the ESM-2 model. (B) Performance binned by the number of MMseqs hits when searching the training set. Larger ESM-2 models perform better at all levels; the 150-million-parameter ESM-2 model is comparable to the 650-million-parameter ESM-1b model. (C) Trajectory of improvement as model scale increases for sequences with different numbers of MMseqs hits. (D) Left-to-right shows models from 8 million to 15 billion parameters, comparing the smaller model (x axis) against the next larger model (y axis) through unsupervised contact precision. Points are PDB proteins colored by change in perplexity for the sequence between the smaller and larger model. Sequences with large changes in contact prediction performance also exhibit large changes in language model understanding measured by perplexity. (E) TM-score on combined CASP14 and CAMEO test sets. Predictions are made by using structure module–only head on top of language models. Points are colored by the change in perplexity between the models. (F) Structure predictions on CAMEO structure 7QQA and CASP target 1056 at all ESM-2 model scales, colored by pLDDT (pink, low; teal, high). For 7QQA, prediction accuracy improves at the 150-million-parameter threshold. For T1056, prediction accuracy improves at the 15-billion-parameter threshold. Credit: Science (2023). DOI: 10.1126/science.ade2574")
Researchers from Facebook AI Research (FAIR) at Meta AI have published a paper in the journal Science detailing a machine-learning-created database of 617 million predicted protein structures. The ESMFold language model described the structures 60 times faster than DeepMinds AlphaFold2, though with less reported accuracy.
The fold predictions were completed in just two weeks on a cluster of about 2,000 GPUs. The initial sequence lengths ranged from 20 to 1,024 nucleotides. 365 million predictions were made with good confidence, and ∼225 million predictions fell within a high confidence of accuracy.
According to the report, "Evolutionary-scale prediction of atomic-level protein structure with a language model," a random sample of 1 million high-confidence results showed that 767,580 proteins have a sequence identity below 90% to any sequence in UniRef90, a database of known protein sequences. Researchers believe this indicates that the proteins are distinct from existing UniRef90 sequences.
The Meta AI team then compared the sample of predicted structures with known structures in the Protein Data Bank, a database for three-dimensional protein structures. At thresholds 0.5 TM-score, 12.6% (125,765 proteins) were without a structural component match. Based on this, researchers estimate that about 28 million proteins (12.6% of 225 million) with high-confidence predictions could characterize regions of protein structure that are distant from existing knowledge.
Predictions based on sequences
A protein begins as a linear sequence of nucleotides copied from DNA (transcription), creating messenger-RNA, a raw ingredient wish list of the protein it will become. The mRNA nucleotides are then translated into amino acids (the raw ingredients). This chain of amino acids then undergoes an incredible transformation into a complex three-dimensional folded shape that, depending on its folded structure, carries out specific intricate cellular functions.
How a protein or enzyme folds in part determines its function because it limits and optimizes what it can interact with. The structure creates an opening or "lock" that only operates with the correct molecular "key." People have been using these lock and key enzymes for everything from the food industry and beer brewing to textiles and biofuel without a detailed understanding of how the proteins are actually folded.
Laundry detergents typically contain several types of enzymes, some of which will be cellulases that break down plant material. When the cellulase enzyme encounters cellulose from a grass stain, the cellulose becomes the key that fits the lock. The enzyme triggers a chemical reaction breaking down the bonds within the grass stain. The same enzyme will do nothing when encountering a lipstick or grease stain, that may be a job for another enzyme.
A single protein enzyme might perform a task thousands or even millions of times per second without breaking, offering industries a low-energy powerhouse of a catalyst and making enzymes an instrumental technology.
Every system in our body also relies on proteins to carry out biological functions. Because the folded structure of a protein is crucial to the activity it can engage in, understanding this structure is critical to understanding how they work when investigating causes of disease.
The ability to predict how a protein will fold based on the primary sequence of amino acids (raw ingredients) would allow medical researchers to better understand protein metabolite interactions and biological functions throughout the body. This higher-resolution understanding could identify hidden disease traits, accelerate research into new or better treatments and somewhat revolutionize modern medicine. Understanding precisely how structure follows the form of raw ingredients (translated mRNA) would also allow researchers to build custom proteins to perform specific tasks in healthcare and industry.
In the decades preceding AI prediction models, scientists modeled the structures of about 190,000 proteins of interest. Machine learning has now generated hundreds of millions of predictions that still need to be confirmed and studied to be useful. While still not reliable enough to replace the slower methodical X-ray crystallography for structure or a controlled assay experiment for function, AI is just getting started. The knowledge gained in the decades to come will likely eclipse everything that came before.
More information: Zeming Lin et al, Evolutionary-scale prediction of atomic-level protein structure with a language model, Science (2023). DOI: 10.1126/science.ade2574
Journal information: Science
© 2023 Science X Network