Concise synthesis of pleurotin developed

From the perspective of chemists, pleurotin is an intriguing molecule.
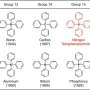
There is strong evidence of untapped therapeutic properties as a tumor inhibitor and antibiotic. It has a fascinating complex structure (six rings! eight stereocenters!). And it has been difficult to synthesize over the decades. The last time chemists pulled that off, the year was 1988 and they needed 26 steps in which to do it.
For Princeton Chemistry's Sorensen Lab, those qualities were part of the attraction for a long-term investment of time and energy that has come to fruition.
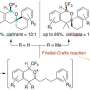
The lab reports a concise synthesis of pleurotin by way of the Diels-Alder reaction and a radical epimerization that flips a cis-hydrindane to the desired trans-hydrindane. Their late-stage intermediate intersects the milestone 1988 synthesis towards the end of the process, thereby reducing the total number of steps needed for the synthesis by thirteen.
The lab's process could yield an expanded family of pleurotin-like anticancer screening candidates which, down the line, may be useful to pharmaceutical companies looking to exploit the promise of pleurotin as a next-generation drug.
"Pleurotin is a very sensitive molecule, it's very reactive. But it hasn't worked out as a drug yet, partly because it's not very water-soluble," said third-year graduate student John Hoskin, lead author on the paper. "Ideally you want to alter its structure: tweak here, change here, put a hydroxy here or a phosphate there, do some very careful modifications."
"And since you can't really do that starting from pleurotin itself, our approach will be to incorporate the changes from a ground-up synthesis, which is only possible because of the brevity of the route. Then you end up with so-called analogs that are very similar to this natural product but that have these strategic changes."
"A Concise Synthesis of Pleurotin Enabled by a Nontraditional C-H Epimerization" was published last month in the Journal of the American Chemical Society by Hoskin, and P.I. Erik Sorensen, the Arthur Allan Patchett Professor in Organic Chemistry in the Department of Chemistry.
"When a chemist looks at a structure like this one, there aren't any obvious strategies that one should take to create it from simple compounds," said Sorensen. His lab first began working on pleurotin in 2008 only to meet with a series of disappointments. Until now.
"If you take pleurotin and say, I want to do site-selective chemistry on its periphery so that we might build new molecules with improved properties, then perhaps there will be better anti-cancer agents," he added. "So John and I were drawn to the challenge of developing a chemical approach for building that framework in as few steps as possible."
"Eight steps is a pretty small number of steps for a molecule of that complexity," said Sorensen. "This research is testimony to John's skill as a designer and executor of organic synthesis."
Untapped promise since 1947
Pleurotin derives from the fungus Pleurotus griseus. Researchers first described the molecule in a paper published in 1947 as inhibiting the growth of Staphylococcus aureus, the source of staph infections. That was 41 years before the milestone synthesis of pleurotin by David Hart, now an emeritus professor at Ohio State University.
But because of the inability to synthesize it easily, pleurotin has not been investigated to its full potential. That's when the Sorensen Lab stepped in.
To pare down the steps toward synthesis, researchers used a tried-and-true tactic in organic synthesis called a 1,5-hydrogen atom transfer, in which a reactive, oxygen-centered radical essentially "reaches over" and plucks a hydrogen off a carbon that is part of the pleurotin structure to make a new radical. Researchers then used that radical to receive hydrogen from an exogenous thiol that would allow for the stereocenter to flip to an alternative—or, trans—configuration.
"We tried a lot of different strategies and finally what ended up working was this inversion step to go from this cis-hydrindane to trans-hydrindane. That's the key insight," said Hoskin. "By utilizing functionality inherent in the molecule—this oxygen—we could, as if we were using a microscopic pair of tweezers, pluck off this hydrogen and flip that carbon to get the needed trans-hydrindane."
The process generates a racemic end-product, making both left- and right-hand versions in equal proportions. Only one of them is likely to be bioactive. Now that the formal synthesis has been complete in a more concise fashion, Hoskin said, the next challenge will be producing just one mirror image version of the molecule, and analogs thereof.
"This research shows the power of a brief synthesis," said Hoskin. "It only takes a week to push through the whole route."
Sorensen added: "I think this work puts us in a favorable position toward our broader aim of expanding the class of pleurotin-based anticancer agents."
More information: Kuo Zhao et al, Contra-Thermodynamic Positional Isomerization of Olefins, Journal of the American Chemical Society (2021). DOI: 10.1021/jacs.1c11681
Journal information: Journal of the American Chemical Society
Provided by Princeton University