Pioneering simulations focus on HIV-1 virus
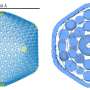
 has been developed using supercomputer resources. (A) Full-view representation of the 150 nm vesicle model. (B) Clipped view of the vesicle and (C) close-up view of the clipped vesicle. CHOL represents cholesterol and the other acronyms represent the 23 different lipids species used in the model. Credit: Bryer et al.")
When is a container not just a container?
For the HIV-1 virus, a double layer of fatty molecules called lipids not only serves as its container, but also plays a key role in the virus's replication and infectivity. Scientists have used supercomputers to complete the first-ever biologically authentic computer model of the HIV-1 virus liposome, its complete spherical lipid bilayer.
What's more, this study comes fresh off the heels of a new atomistic model of the HIV-1 capsid, which contains its genetic material. The scientists are hopeful this basic research into viral envelopes can help efforts to develop new HIV-1 therapeutics, as well as laying a foundation for study of other enveloped viruses such as the novel coronavirus, SARS-CoV-2.
"This work represents an investigation of the HIV-1 liposome at full-scale, and with an unprecedented level of chemical complexity," said Alex Bryer, a Ph.D. student in the Perilla Laboratory, Department of Chemistry and Biochemistry, University of Delaware. Bryer is the lead author of the liposome-modeling research, published January 2022 in the journal PLOS Computational Biology.
The science team developed a complex chemical model of the HIV-1 liposome that revealed key characteristics of the liposome's asymmetry. Most such models assume a geometrically uniform structure and don't capture the asymmetry inherent in such biological containers.
Lipid flip-flop
Bryer and his co-authors investigated a mechanism that's known colloquially as "lipid flip-flop," which is when lipids in one of the leaflets of the bilayer are moved or transported to the other leaflet. The leaflets flip-flop the lipids and exchange the molecules for various purposes such as achieving a dynamic equilibrium.
"For the spherical vesicle model of the liposome, our simulations show that asymmetry occurs spontaneously even without embedded proteins, and the vesicle can flip-flop to maintain an asymmetric composition within tight tolerances—even over biological timescales in excess of five microseconds," Bryer said.
Interestingly, the science team did not observe incidence of flip-flop in a flat membrane system, which suggests that curvature of the envelope is intimately related to this biological process.
"Nothing like this has ever been simulated before." said study co-author Juan R. Perilla, an assistant professor in the Department of Chemistry and Biochemistry, University of Delaware.
"What was surprising for us is this dynamic equilibrium that the vesicle shows," Perilla added. "Lipids are moving in and out, but the overall composition is not changing—that was surprising."
Key asymmetryThis key finding shows that the complex, asymmetric membrane composition of the HIV-1 virus can lead to macroscopic properties such as the differential displacement between leaflets and lipid microdomain formation.
That formation might have implications in how membrane proteins, which often localize within specific lipid microdomains, interact with the membrane and carry out functions such as binding to host cells and allowing the virus to enter them.
For HIV-1, it's known that microdomains form and act as a target for the localization of membrane proteins. One protein in particular, gp41, is critical for membrane fusion, which is the process of HIV-1 joining with the host cell membrane and ultimately infecting it.
"It's thought that gp41 localizes to these domains," Bryer said. "What we showed was that these microdomains can form in the vesicle without the aid of proteins. They seem to emerge spontaneously."
This finding might also explain the preferential budding behavior in HIV-1 viral replication, without the need of embedded proteins in mediating the formation of the microdomains that enable budding.
Supercomputer simulations
The computer model Bryer and colleagues developed is 150 nm in diameter and consists of 24 different chemical constituents. There are more than 300,000 total lipid molecules, solvated in water and ionized with sodium chloride, to represent a biological environment. The science team employed a coarse-grained model known as MARTINI, which allowed them to reduce the degrees of freedom in the molecular system and achieve simulation sampling over microsecond timescales.
The scientists were awarded supercomputer allocations and training by XSEDE funded by the National Science Foundation. Through XSEDE, they used the Stampede2 system at the Texas Advanced Computing Center (TACC) and Bridges-2 at the Pittsburgh Supercomputing Center (PSC). Additionally, they used Grizzly at the Los Alamos National Laboratory; Blue Waters at the National Center for Supercomputing Applications; and the Frontera system at TACC.
"Our study wouldn't have been possible without XSEDE resources," Bryer said. "We can achieve some very high sampling efficiencies using Stampede2 Skylake nodes, both to run the simulations and perform analyses."
"I was able to perform calculations, and without needing to transfer data, I could set up a visualization session through the TACC portal and analyze and work with my data directly on Stampede2. That's amazing," added Bryer. He found that not having to transfer terabytes of data into a separate visualization computer node was "just huge in terms of productivity.""We also used quite a bit of the high memory nodes on Bridges-2 of PSC," Perilla said. They helped power simulations that compared the control, a flat HIV-1 viral membrane, to the curved one in dynamic equilibrium.
What's more, the Perilla Lab has transferred the simulation work to their local cluster, the XSEDE-allocated DARWIN system of the University of Delaware.
"It's important to highlight the fact that XSEDE does not just provide resources, which are extremely valuable. There's training and other opportunities such as workshops," Perilla said.
"When I joined the group, I had never logged into a supercomputer," Bryer said. He recalled valuable training in XSEDE workshops on OpenMP, MPI, and OpenACC, which assist scientists in parallelizing their computer code.
Frontera work
Bryer also highlighted the analysis work run on TACC's Frontera, the fastest academic supercomputer in the world. "Parallel I/O via Luster is what made a lot of the analyses possible in the manuscript," Bryer said. "On Frontera we were able to classify the volume surrounding the vesicle quickly and process our data in minutes. We estimated it might take about three weeks if we were to run the analysis in a serial naive implementation."
The Perilla Lab has focused all of this computing power and expertise into learning more about the mysteries of what happens to the HIV-1 viral envelope during infection.
"While this study does not provide the whole answer, it's getting there in what the lipids are doing and what integral membrane proteins are doing or could be doing; and not only how proteins like gp41 interact with human receptors but also how they transmit their signals and how that is related to lipid composition," Perilla said.
"This computational study provides an opportunity for drug development research," Perilla added.
Since lipid symmetry is maintained by the curvature of the envelope, a promising possibility yet unexplored is development of small molecules that affect the symmetry and potentially yield a therapeutic target.
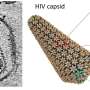
HIV-1 capsid
Just prior to the liposome research, Perilla and colleagues also broke new ground in using supercomputers to build the first-ever atomistic model of the HIV-1 capsid, the envelope for its genetic material, in the presence of the metabolite IP6. The work was published November 2021 in the journal Science Advances. It also used the Bridges-2 and Stampede2 supercomputers allocated through XSEDE.
The simulations, validated by cryo-electron tomography data, showed that IP6 was able to bind in two locations to the capsid, instead of just one as previously thought. This finding is important because during infection, the capsid is exposed to the cytoplasm and has to go through the nuclear import mechanism, namely the nuclear pore complex. All these pieces together point to the capsid being able to "sense" in an as yet unknown way the concentration of IP6.
Said Perilla: "Computationally, these are very unique simulations because of the number of degrees of freedom involved. Nobody's ever walked this path before. We're walking through the dark. And we're making tools that can help us see beyond where we are."
More information: Alexander J. Bryer et al, Full scale structural, mechanical and dynamical properties of HIV-1 liposomes, PLOS Computational Biology (2022). DOI: 10.1371/journal.pcbi.1009781
Journal information: PLoS Computational Biology , Science Advances
Provided by University of Texas at Austin