Probing how proteins pair up inside cells
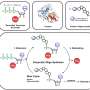
 use flanking regions outside their core amino acid sequence to recognize proteins such as ENAH (gray). Credit: Theresa Hwang")
Despite its minute size, a single cell contains billions of molecules that bustle around and bind to one another, carrying out vital functions. The human genome encodes about 20,000 proteins, most of which interact with partner proteins to mediate upwards of 400,000 distinct interactions. These partners don't just latch onto one another haphazardly; they only bind to very specific companions that they must recognize inside the crowded cell. If they create the wrong pairings—or even the right pairings at the wrong place or wrong time—cancer or other diseases can ensue. Scientists are hard at work investigating these protein-protein relationships, in order to understand how they work, and potentially create drugs that disrupt or mimic them to treat disease.
The average human protein is composed of approximately 400 building blocks called amino acids, which are strung together and folded into a complex 3D structure. Within this long string of building blocks, some proteins contain stretches of 4-6 amino acids called short linear motifs (SLiMs), which mediate protein-protein interactions. Despite their simplicity and small size, SLiMs and their binding partners facilitate key cellular processes. However, it's been historically difficult to devise experiments to probe how SLiMs recognize their specific binding partners.
To address this problem, a group led by Theresa Hwang Ph.D. '21 designed a screening method to understand how SLiMs selectively bind to certain proteins, and even distinguish between those with similar structures. Using the detailed information they gleaned from studying these interactions, the researchers created their own synthetic molecule capable of binding extremely tightly to a protein called ENAH, which is implicated in cancer metastasis. The team shared their findings in a pair of eLife studies, one published on January 25, 2022 and the other on December 2, 2021.
"The ability to test hundreds of thousands of potential SLiMs for binding provides a powerful tool to explore why proteins prefer specific SLiM partners over others," says Amy Keating, professor of biology and biological engineering and the senior author on both studies. "As we gain an understanding of the tricks that a protein uses to select its partners, we can apply these in protein design to make our own binders to modulate protein function for research or therapeutic purposes."
Most existing screens for SLiMs simply select for short, tight binders, while neglecting SLiMs that don't grip their partner proteins quite as strongly. To survey SLiMs with a wide range of binding affinities, Keating, Hwang, and their colleagues developed their own screen called MassTitr.
The researchers also suspected that the amino acids on either side of the SLiM's core 4-6 amino acid sequence might play an underappreciated role in binding. To test their theory, they used MassTitr to screen the human proteome in longer chunks comprised of 36 amino acids, in order to see which "extended" SLiMs would associate with the protein ENAH.
ENAH, sometimes referred to as Mena, helps cells to move. This ability to migrate is critical for healthy cells, but cancer cells can coopt it to spread. Scientists have found that reducing the amount of ENAH decreases the cancer cells's ability to invade other tissues—suggesting that formulating drugs to disrupt this protein and its interactions could treat cancer.
Thanks to MassTitr, the team identified 33 SLiM-containing proteins that bound to ENAH—19 of which are potentially novel binding partners. They also discovered three distinct patterns of amino acids flanking core SLiM sequences that helped the SLiMs bind even tighter to ENAH. Of these extended SLiMs, one found in a protein called PCARE bound to ENAH with the highest known affinity of any SLiM to date.
Next, the researchers combined a computer program called dTERMen with X-ray crystallography in order understand how and why PCARE binds to ENAH over ENAH's two nearly identical sister proteins (VASP and EVL). Hwang and her colleagues saw that the amino acids flanking PCARE's core SliM caused ENAH to change shape slightly when the two made contact, allowing the binding sites to latch onto one another. VASP and EVL, by contrast, could not undergo this structural change, so the PCARE SliM did not bind to either of them as tightly.
Inspired by this unique interaction, Hwang designed her own protein that bound to ENAH with unprecedented affinity and specificity. "It was exciting that we were able to come up with such a specific binder," she says. "This work lays the foundation for designing synthetic molecules with the potential to disrupt protein-protein interactions that cause disease—or to help scientists learn more about ENAH and other SLiM-binding proteins."
Ylva Ivarsson, a professor of biochemistry at Uppsala University who was not involved with the study, says that understanding how proteins find their binding partners is a question of fundamental importance to cell function and regulation. The two eLife studies, she explains, show that extended SLiMs play an underappreciated role in determining the affinity and specificity of these binding interactions.
"The studies shed light on the idea that context matters, and provide a screening strategy for a variety of context-dependent binding interactions," she says. "Hwang and co-authors have created valuable tools for dissecting the cellular function of proteins and their binding partners. Their approach could even inspire ENAH-specific inhibitors for therapeutic purposes."
Hwang's biggest takeaway from the project is that things are not always as they seem: even short, simple protein segments can play complex roles in the cell. As she puts it: "We should really appreciate SLiMs more."
More information: Native proline-rich motifs exploit sequence context to target actin-remodeling Ena/VASP protein ENAH, eLife, DOI: 10.7554/eLife.70680 , elifesciences.org/articles/70680
A distributed residue network permits conformational binding specificity in a conserved family of actin remodelers, eLife, DOI: 10.7554/eLife.70601 , elifesciences.org/articles/70601
Journal information: eLife
Provided by Massachusetts Institute of Technology Department of Biology