New computer modelling could boost drug discovery
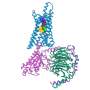
 The overall view of the receptors with a bound allosteric modulator. (A) The M2 receptor bound to LY2119620 (green), a PAM binding at the extracellular side, and the orthosteric ligand, Iperoxo (blue). (B) The β2 receptor bound to Cmp-15, a NAM, at the intracellular side. (C) The P2Y1 receptor in complex with BPTU, a NAM bound at the lipid interface. The receptors are in wild type with the rebuilt short intracellular loop 3 (ICL3) fragment. (D–F) The binding interactions between the allosteric modulator and the receptor obtained from MD simulations of the X-ray receptor–ligand complexes for the M2, β2, and P2Y1 receptors. The key residues forming strong interactions and the allosteric ligands are in gray and green sticks, respectively. Hydrogen bonds and π–π interactions are shown as pink and cyan dashed lines, respectively. Credit: DOI: 10.1021/acscentsci.1c00802")
Scientists from Queen's University Belfast have developed a computer-aided data tool that could improve treatment for a range of illnesses.
The computer modeling tool will predict novel sites of binding for potential drugs that are more selective, leading to more effective drug targeting, increasing therapeutic efficacy and reducing side effects.
The data tool or protocol will uncover a novel class of compounds—allosteric drugs in G protein-coupled receptors (GPCRs).
GPCRs are the largest membrane protein family that transduce a signal inside cells from hormones, neurotransmitters, and other endogenous molecules. As a result of their broad influence on human physiology, GPCRs are drug targets in many therapeutic areas such as inflammation, infertility, metabolic and neurological disorders, viral infections and cancer. Currently over a third of drugs act via GPCRs. Despite the substantial therapeutic success, the discovery of GPCR drugs is challenging due to promiscuous binding and subsequent side effects.
Recent studies point to the existence of other binding sites, called allosteric sites that drugs can bind to and provide several therapeutic benefits. However, the discovery of allosteric sites and drugs has been mostly serendipitous. Recent X-ray crystallography, that determines the atomic and molecular structure, and cryo-electron microscopy that offers 3D models of several GPCRs offer opportunities to develop computer-aided methodologies to search for allosteric sites.
The researchers developed a computer-aided protocol to map allosteric sites in GPCRs with a view to start rational search of allosteric drugs, presenting the opportunity for new solutions and therapies for a range of diseases.
Dr. Irina Tikhonova from the School of Pharmacy at Queen's University and senior author, explains: "We have developed a novel, cost-effective and rapid pipeline for the discovery of GPCRs allosteric sites, which overcomes the limitations of current computational protocols such as membrane distortion and non-specific binding.
"Our pipeline can identify allosteric sites in a short time, which makes it suitable for industry settings. As such, our pipeline is a feasible solution to initiate structure-based search of allosteric drugs for any membrane-bound drug targets that have an impact on cancer, inflammation, and CNS diseases."
This research published in ACS Central Science is a collaboration with Queen's University Belfast and Queen Mary University of London.
More information: Antonella Ciancetta et al, Probe Confined Dynamic Mapping for G Protein-Coupled Receptor Allosteric Site Prediction, ACS Central Science (2021). DOI: 10.1021/acscentsci.1c00802
Journal information: ACS Central Science
Provided by Queen's University Belfast