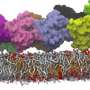
A general-purpose force field for coarse-grained molecular dynamics

Simulating the interactions between atoms and molecules is important for many scientific studies. However, accurate simulations can take a long time, which limits their use. To speed up simulations without sacrificing too much detail, Siewert-Jan Marrink, Professor of Molecular Dynamics at the University of Groningen, designed a set of parameters that allow fast but accurate coarse-grained simulations. In a paper that was published on 29 March in Nature Methods, Marrink and his co-workers present a third release of what is known as the Martini force field.
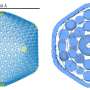
"Our Martini force field typically combines four heavy atoms and any attached protons into one so-called bead. Users can then simulate interactions between the beads, which reduces the number of particles in the simulation," Marrink explains. The reduction in particles reduces the computing time by up to three orders of magnitude. "This allows you to simulate longer processes, over a period of milliseconds rather than microseconds, or to simulate bigger or more molecules."
The Martini force field—named after Martin of Tours, the city of Groningen's patron saint and name-giver to its biggest church, but with a firm nod to the famous cocktail—has been in use for well over a decade and was first developed by Marrink to simulate lipid membranes. "The first scientific paper that mentions the Martini force field dates from 2007, when the second release appeared." It has gathered over four thousand citations so far.
A force field is a set of parameters for use in a molecular dynamics simulation program. The accuracy of the result depends on how the parameters that define the interactions between the beads are determined. In the new version, these interactions have been recalibrated with many more reference data than the previous release. "By using the force field ourselves and through the feedback that we received from users, we discovered where improvements could be made," explains Marrink. A lot of feedback reaches him through the active user platform that his research group maintains online.
The first author of the paper, Dr. Paulo C.T. Souza, who is a postdoctoral researcher in Marrink's group, has worked on the recalibration of the parameters for four years. "For this job, you need to understand how the force field is put together but you also need biological and chemical intuitions and a good background in physics. Paulo has all of that."
The new version is a generic force field that can be used for simulations of soft matter, such as lipid membranes, proteins, polymers and DNA. "The original force field was developed for biological molecules but it is also increasingly often used in materials science." Simulations that are based on the Martini force field are used to explain experimental results, predict interactions and emergent behavior between molecules and as a high-throughput system to evaluate, for example, the interactions of drug leads to binding sites. Just like with its predecessor, it is possible to backmap results from Martini 3-based simulations to atomistic simulations. "This allows you to zoom in on interesting interactions."
The third release of Martini can be downloaded for free from the Martini website. "We want this force field to be available to everyone in the research community," says Marrink. "Putting it behind a paywall would have been possible but that is not what I want. Martini was developed as an academic project, not a commercial one."
The upgrade to Martini 3 took about 10 years to complete. And Marrink and his co-workers are eager to explore its potential. "The next step in our force field-related research is to further improve the models for biomolecules in general, such as lipids, proteins, sugars and nucleotides, using the new Martini 3 parameters. Our ultimate goal is being able to simulate an entire cell at a molecular resolution within the next five years."
A future development is to include chemical reactions in the force field. "Chemical reactions change the nature of molecules but are not incorporated into existing force fields. Reactions require quantum mechanical calculations, which take a lot of time. We want to make a faster, coarse-grained alternative." A final item on the wish list for Martini 4 is the inclusion of changes in acidity.
But for now, Marrink is enjoying the launch of Martini 3 and is interested to see how it is going to affect the modeling community. "We expect that it will certainly impact fundamental science in many areas, from biophysics to drug development and materials science."
More information: Martini 3: a general purpose force field for coarse-grained molecular dynamics, Nature Methods (2021). DOI: 10.1038/s41592-021-01098-3
Martini force field website and user community: http://cgmartini.nl/
Journal information: Nature Methods
Provided by University of Groningen