First simulation of a full-sized mitochondrial membrane

Scientists from the University of Groningen have developed a method that combines different resolution levels in a computer simulation of biological membranes. Their algorithm backmaps a large-scale model that includes features such as membrane curvature to its corresponding coarse-grained molecular model. This has allowed them to zoom in on toxin-induced membrane budding and to simulate a full-sized mitochondrial lipid membrane. Their approach, which was published in the journal Nature Communications on 8 May, opens the way to whole-cell simulations at a molecular level.
Molecular dynamics simulations are a powerful tool to study the movements and interactions of atoms and molecules. However, in many biological processes, large-scale changes in, for example, membrane shape are important. "These shape changes are of fundamental importance to the cell's functioning," explains Siewert-Jan Marrink, Professor of Molecular Dynamics at the University of Groningen. "However, the time and length scale of these membrane shape changes are too large for simulations at a molecular resolution."
Challenging
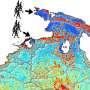
Even though an increase in computing power allows more complex and longer simulations, cell structures such as mitochondria are still beyond reach. That is why the Molecular Dynamics group has developed an algorithm that links large-scale changes to molecular level simulations. For mitochondria, they started with an electron micrograph density map. The densities were translated into lipid structures and these were used as the input for a molecular dynamics simulation with the Coarse Grain (CG) Martini force field, previously developed by Marrink.

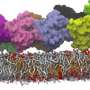
"The difficult part is to place the lipids in the correct orientation in this density map, which is especially challenging in bent areas," adds Wria Pezeshkian, a postdoctoral researcher in Marrink's team and co-author of the paper. The algorithm allows users to add different kinds of lipids to the membrane, at a realistic packing level. Using this approach, Marrink and his colleagues were able to simulate the entire lipid membrane of a mitochondrion for two nanoseconds. Pezeshkian: "This structure contained more than five million lipids, which meant that the simulation had to deal with 80 million particles as each lipid molecule consists of multiple particles."
Triangles
Considering the size and shape, this simulation's complexity is larger than any simulation performed previously. "A simulation of microseconds would have been possible but, as we had no information on the localization of the proteins in the mitochondrial membrane, it only contained lipids and is therefore unstable," explains Marrink. Adding this extra complexity to the simulation is certainly possible and is currently in progress.
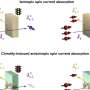
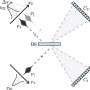
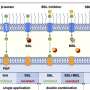
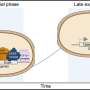
Instead of a density map, the input for the system could also be a continuum model, which represents the membrane surface as triangles made up of nodes that are connected by 'springs." Such a model can calculate forces generated by membrane deformation. Backmapping lipids and toxin proteins onto the corresponding parts of this model allowed Marrink and his colleagues to zoom in on molecular behavior in the stalk of a membrane bud that was induced by the joint action of many toxins.
Synthetic cell
"Our final goal is to simulate an entire eukaryotic cell and zoom in on specific parts of this object," says Marrink. This is currently out of reach, although the current system already allows simulation of large objects inside a cell, such as the endoplasmic reticulum or the Golgi apparatus. "And we could probably simulate a red blood cell."
A simple synthetic cell may soon be within reach. Marrink is involved in a project aimed at creating a synthetic cell and being able to simulate processes such as cell division would help its design. "We would really like to know which lipids and proteins could play a role in cell constriction during division."
More information: Weria Pezeshkian, Melanie König, Tsjerk A. Wassenaar and Siewert J. Marrink: Backmapping triangulated surfaces to coarse-grained membrane models. Nature Communications (2020). DOI: 10.1038/s41467-020-16094-y
Journal information: Nature Communications
Provided by University of Groningen