How does a cell regulate the repair of its damaged DNA?

Human DNA can be modified or damaged in a variety of ways that can alter its sequence and potentially change the information that it encodes, from UV radiation to chemicals in our own bodies. One way the body can accurately copy damaged DNA is through a process called translesion DNA synthesis, which involves a complex of proteins called Rad6/Rad18. Assistant Professor of Chemistry Mark Hedglin investigated how the Rad6/Rad18 protein complex moves along filaments produced by a type of DNA replication factor during translesion DNA synthesis. This new insight could have important implications for cancer treatments, as dysfunction of this protein complex has been linked to several cancers and complications with chemotherapy.
A paper describing the results was recently published in the journal Biochemistry. We talked to Hedglin about this research:
Q: How does DNA become damaged?
Hedglin: Our genetic information (i.e., genome) is encoded in strands of DNA that must be accurately replicated and passed on every time a cell divides. DNA is constantly exposed to things that can chemically modify DNA. For example, reactive metabolites produced in our own bodies and environmental toxicants, such as ultraviolet radiation from sunlight, can chemically modify, or damage, DNA. Although this type of damage is relatively infrequent throughout our genome, it can alter the coding properties of DNA, leading to the generation of incorrect copies of our genome that are riddled with errors (i.e., mutations).
Q: How can a cell deal with DNA damage?
Hedglin: Numerous cellular pathways can repair damaged DNA before it is copied but these pathways are not 100% efficient, and many of the modifications escape detection. To account for this, our cells use specialized pathways, such as translesion DNA synthesis (TLS), that can accurately replicate DNA that has been damaged by chemical modification.
Q: What was your motivation for this study?
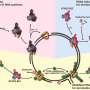
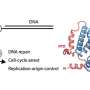
Hedgiln: For translesion DNA synthesis to occur, the Rad6/Rad18 complex must find and alter an essential DNA replication factor called PCNA that resides at DNA damage sites. Keep in mind, PCNA is highly abundant in our cells but DNA damage is relatively rare throughout our genome, so the proportion of the cellular PCNA stock that resides at sites of DNA damage is extremely small (< 10%). One of the burning questions in the field that has long remained unanswered was "how can Rad6/Rad18 complexes locate PCNA at DNA damage sites as opposed to anywhere else along the genome?" It was previously suggested that Rad6/Rad18 is recruited to a site of DNA damage by another essential DNA replication factor, RPA, that accumulates at these sites and forms long, extended filaments next to the PCNA. As scientists interested in enzyme mechanisms, we were highly intrigued by how the recruitment of a Rad6/Rad18 complex to an RPA filament at a site of DNA damage ultimately allowed TLS to occur.
Simply put, a Rad6/Rad18 complex floating around in solution most likely engages an RPA molecule along the filament that is far away from the PCNA, so how does the Rad6/Rad18 complex ultimately locate the PCNA? There wasn't any evidence hinting at a given mechanism and many, many models could be envisioned but they were all just speculation. It was a really exciting question to address because we really had no idea what the answer would be.
Q: What were the main results of this study?
Hedglin: What we found is that RPA is indeed required to recruit a Rad6/Rad18 complex to a DNA damage site and that once a Rad6/Rad18 complex is engaged with an RPA filament, it randomly moves along the filament by transferring between adjacent RPA molecules until it locates PCNA.
Q: Why is this important?
Hedglin: This discovery was unexpected and quite astounding for many reasons. First, to the best of our knowledge, this is the first example of a protein complex moving along a protein filament independently of an energy source. Typically, diffusion of proteins or protein complexes along protein filaments is directional (either one direction or the other, not both) and driven by an energy source (like ATP hydrolysis). We found that the Rad6/Rad18 complex randomly moves along RPA filaments by thermally-driven diffusion. These studies alter our fundamental understanding of the dynamics of protein-protein interactions. Second, our findings addressed a large gap in the catalytic mechanism for the Rad6/Rad18 complex; how does the recruitment a Rad6/Rad18 complex to an RPA filament ultimately lead to TLS? Dysfunction of the Rad6/Rad18 complex has been linked to many cancers as well as resistance to many common chemotherapies. Thus, a more comprehensive picture of the catalytic mechanism for the Rad6/Rad18 complex can identify dysfunctions that promote cancer and inspire fresh approaches for targeting the Rad6/Rad18 complex with chemotherapeutics. Finally, the translocation behavior of the Rad6/Rad18 complex that we discovered offered a sound explanation that accounted for many obvious challenges to the ability of Rad6/Rad18 complexes to alter PCNA in vivo. These challenges are addressed in detail in the manuscript.
More information: Mingjie Li et al. PCNA Monoubiquitination Is Regulated by Diffusion of Rad6/Rad18 Complexes along RPA Filaments, Biochemistry (2020). DOI: 10.1021/acs.biochem.0c00849
Journal information: Biochemistry
Provided by Pennsylvania State University