Active learning accelerates redox-flow battery discovery

By using active learning, scientists are more quickly finding suitable candidates for redox-flow batteries.
When it comes time to design a new battery chemistry, scientists only can try a handful of possibilities experimentally, as it takes time and resources to synthesize and investigate each new molecule. By performing reliable molecular simulations using supercomputers, researchers can speed up the desired materials screening process and expand the breadth of their search, while getting detailed information about the possibilities inherent in different chemistries.
However, even high-throughput simulations run on these supercomputers can only look at a fraction of the possible viable chemistries that exist for certain types of batteries. In a new study from the U.S. Department of Energy's (DOE) Argonne National Laboratory, researchers are taking the next step in accelerating the hunt for the best possible battery components by employing artificial intelligence.
The study team, led by Argonne chemist Rajeev Surendran Assary, investigated the inner workings of redox flow batteries, in which chemical energy is stored in dissolved molecules that interact with electrodes. Flow batteries are promising for applications in the electric grid. They replace solid cathodes and anodes with liquid solutions infused with molecules that store and release energy. Conventional flow batteries are based on molecules that have one charge storing element per molecule, with limited versatility. Researchers at the Joint Center for Energy Storage Research (JCESR), a DOE Energy Innovation Hub led by Argonne, introduced the concept of storing and releasing energy with materials called "redox active polymers," or redoxmers, which are based on larger molecules, each with tens of charge storing elements.
Compared to conventional systems, redoxmers allow much greater flexibility to independently customize many aspects of battery characteristics and performance. Redoxmer flow batteries open a new door on flow battery design because they can provide high functionality at low cost, with little harm to the environment. JCESR's redoxmer flow batteries have the potential to transform how we think about and use flow batteries for the grid.
In the case of the redoxmers under study, Assary and his colleagues noticed that, as the battery charges and discharges, they tend to form an inactive film. To prevent this phenomenon, the Argonne team looked to design a redoxmer that could be electrically cleaved at a particular voltage, freeing it to re-enter the electrolyte solution.
"You can think of it like cleaning a pan that you cook on," said Argonne postdoctoral researcher Hieu Doan, another author of the study. "To remove sticky food residues more easily, you may use high heat, and that's what we're doing with electricity."
The researchers wanted to have the cleaving voltage be just outside the battery's normal operating window, so that it wouldn't interfere with performance, but also wouldn't require a lot of additional energy.
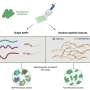
To find a redoxmer that would cleave at the appropriate voltage, Assary and the team turned to Argonne's Bebop supercomputer at the Laboratory Computing Resource Center. First, the researchers ran a set of 1,400 different redoxmers using density functional theory (DFT) calculations, which are highly accurate but computationally expensive. However, these 1,400 redoxmers represented only a tiny slice of the total chemical space that the researchers were interested in.
"Experimentally, it may take months to synthesize and test a dozen of these redoxmers, so to be able to study more than a thousand redoxmers on the computer in detail is essential," Assary said.
Each of these redoxmers consists of a molecular scaffold on which are placed a variety of different chemical functional groups—which are additional atoms or molecules. "The scaffold was designed based on suggestions from our experimental collaborators," Doan said. While the scaffold is consistent across the redoxmers, varying the functional groups gives different properties.
To find the ideal molecules from a larger dataset consisting of more than 100,000 redoxmers without running extensive DFT calculations, the researchers used a machine learning technique called active learning. This larger dataset included redoxmers that were structurally similar to those in the original DFT dataset of 1,400 molecules—in so far as both sets of molecules used the same scaffold. However, because of the different ways the functional groups were populated, the properties diverged.
"How much learning you can do in machine learning is limited by your training dataset," Assary said. "You can only know what you've seen, and if you have something different that you're trying to make predictions about, it may not be effective."
Rather than training on the entirety of the data, Assary and his colleagues trained the model on only a handful of different redoxmer possibilities. According to Doan, after training the model with 10 data points, the model picks the 11th data point on its own from the remaining data pool.
"The model guarantees that by adding this new data point to the training set, it will become better, and then we can train it again," Doan said. "Whatever maximizes the accuracy of the model, that will be the next data point to pick."
Assary said that to identify 30 molecules with the desired properties from an initial dataset of 1,400, only took 70 picks. With random picking, only 9 percent of picks would have been successful, representing a fivefold improvement.
"An improvement this substantial over such a large chemical space is remarkable," Assary said. Indeed, when the same approach was applied to the 100,000+ dataset, it successfully found 42 desired molecules within 100 picks.
A paper based on the study, "Quantum chemistry-informed active learning to accelerate the design and discovery of sustainable energy storage materials," was published in the May 28 issue of Chemistry of Materials.
More information: Hieu A. Doan et al. Quantum Chemistry-Informed Active Learning to Accelerate the Design and Discovery of Sustainable Energy Storage Materials, Chemistry of Materials (2020). DOI: 10.1021/acs.chemmater.0c00768
Journal information: Chemistry of Materials
Provided by Argonne National Laboratory