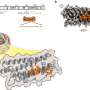
Coronavirus binding and core helix fusion simulated on supercomputer
 in its spike protein that latch on to its host cell, leading to infection. A computer model is simulating these dynamics on Longhorn, the subsystem of the Frontera supercomputer at the Texas Advanced Computing Center (TACC). Credit: Numan Oezguen, Texas Children's Hospital and Baylor College of Medicine.")
Like the mythical Jacob's Ladder that reaches for the heavens, the coronavirus carries its own far-reaching molecular ladder-like apparatus. The coronavirus essentially builds a ladder to its host cell by piecing together and extending three sections in the core of the spike proteins adorning its surface.
The virus infects its host cell by first binding one of its spike proteins and then fusing its helical core to the host cell. Scientists don't yet fully understand the details of how the coronavirus binds and fuses. A computer model is simulating these dynamics on Longhorn, the subsystem of the Frontera supercomputer at the Texas Advanced Computing Center (TACC).
"Our work is basically trying to understand how the virus works," said Numan Oezguen, an instructor at the Microbiome Center of Texas Children's Hospital and also at Baylor College of Medicine. "Once we have this understanding, then we can devise strategies to prevent it from infecting and getting us sick."
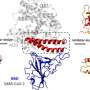
Oezguen's research focuses on the coronavirus spike protein, a molecule made of three identical parts bound to the protein envelope of the virus. It takes its first step in infection by changing its shape, or conformation, so that it can bind to the ACE2 receptor typically found in high numbers in the lungs.
"This is basically one of the first questions that I would like to answer with this project that I'm currently running," Oezguen said. "Can I see when and how the receptor binding domain of the virus that binds to the ACE2 changes its conformation? How does it move from its closed, or down, conformation into the up conformation that is capable of binding to the receptor? And can we disable it?"
Scientists discovered the coronavirus that causes COVID-19 in late 2019. Luckily, the structure and the binding mode of the spike molecule is almost the same as that of the virus that caused the 2003 SARS outbreak. This similarity has helped Oezguen develop his model of SARS-CoV-2 spike. The cryo-EM structure of the novel coronavirus spike protein had many gaps that had to be filled in order to achieve an atomistic detailed model suitable for simulations. With this in place, he put his model of a half-million atoms to the test to answer questions such as how far the spike protein moves, how fast, and what triggers its movement.

Oezguen took his coronavirus model to the Longhorn subsystem of the Frontera supercomputer, where, since April of 2020, he's completed over 47,000 node hours of simulations. The work is ongoing. Longhorn utilizes multiple graphics processing units (GPUs) and supports AMBER18, the molecular dynamics program that he uses. He uses two GPUs in his simulations, each bearing 5,120 processing cores. "This is a huge advantage when it comes to these simulations," Oezguen said.
"I'm very happy with Longhorn. Even the very large system with half a million atoms progresses at about 20 million femtoseconds per day. I'm calculating every femtosecond the position and velocity of each atom. This is mind-boggling, actually, how much computation is going on," Oezguen explained.
He credited Longhorn's speed to its ability to run multiple GPUs on the same node, which eliminates the communication overhead between nodes.
"Even with these fast resources," he continued, "the goal for one microsecond has taken about 50 days. Imagine if you would have less efficient machines to work with. It will take so long you would not be able to finish it in a human lifetime. We've come very far, and I'm very grateful to have access to this great resource," Oezguen said.
Oezguen successfully proposed his research exploring the dynamics of the SARS-CoV-2 spike protein to the COVID-19 High Performance Computing Consortium. Dozens of national and international supercomputing facilities, industry and organizations (including TACC) have volunteered their resources to the consortium in support of scientists' effort to combat the coronavirus.
Oezguen has completed his first set of simulations of the spike changing conformation to bind to the ACE2 receptor of the host cell. The analysis isn't complete, but he hasn't yet seen the expected movement of the receptor binding domain of the virus changing from the down to the up position, indicating that it is primed for infection.
"Hopefully, I will see the movement and then analyze what regions of the protein are moving first or enabling the movement. Once we know this, then we can think about ways of preventing this. If we can prevent the up movement of the receptor binding domain on the spike, then everything else stops. The virus cannot enter the cell. This is crucial," said Oezguen.
Another possibility he brought up was that the environmental conditions might be throwing a monkey wrench into the simulations. "It seems that the environmental pH conditions need to be acidic. This is absolutely counterintuitive," Oezguen said. That's because blood pH is typically slightly basic, right where Oezguen put his simulations.
"With the next set of simulations that I'm setting up, I'm trying to see whether this movement is going to happen on a faster timescale or happen at all in an acidic environment. After this question is answered, then I can go further on and see whether we can interfere with it," Oezguen said.
And if the coronavirus does bind, what comes next? Scientists know from experimental work that the spike protein needs to be cleaved by a host protease so that its actual entry machinery can be exposed. This is where the coronavirus' ladder-like helical domains get exposed and attach to the host membrane.
"The virus spike core helices also need to change their conformations very dramatically," Oezguen said. "If you look at animations of how this helix coil bundle changes its conformation, you would be amazed." That's because from its initial packed conformation, the ladder appendage extends its length by a factor of three or four, by rearrangements of the core helices, so that it can reach the host cell and start the fusion process.
"In this simulation, what I am using is a very appropriate tool to study this kind of structural changes. That's why I'm setting a second set of molecular dynamics simulations exactly to test this," Oezguen said. The second set doesn't simulate the whole spike protein, but only the half of the spike that is exposed to the solvent after cleavage. This basic research gives scientists the potential to interfere with the coronavirus.
Said Oezguen: "If we can stop these conformational changes, we will win the race against the virus. Because if it cannot change this conformation, or it cannot do it efficiently, then it cannot fuse with the host membrane and therefore enter the cell. Generating the knowledge of how the virus is actually functioning forms the basis for any intervention that can later on be devised and then applied."
Provided by Texas Advanced Computing Center