New approach to metabolomics research could prove game changer

Accurate identification of metabolites, and other small chemicals, in biological and environmental samples has historically fallen short when using traditional methods. Conventional tactics rely on pure reference compounds, called standards, to recognize the same molecules in complex samples. These approaches are limited by the availability of the pure chemicals that are used as the standards.
"We really wanted to bypass the current paradigm of how a metabolomics experiment is conducted and how molecules are confidently identified," said Tom Metz, biomedical scientist at Pacific Northwest National Laboratory (PNNL) and director of the Pacific Northwest Advanced Compound Identification Core.
One problem with the current method is that there are only so many pure compounds researchers can purchase from suppliers; most suppliers have access to around 3,000–4,000 compounds.
"If you consider what's predicted to occur in nature, you're looking at >1030 compounds or more that could be possible," said Metz. "So, when you compare the few thousand standard chemicals you have access to against the vast number of potential compounds, you're not even close."
Standards-free identification approach
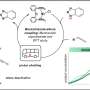
To solve this problem Metz and his team at PNNL conceptualized an approach—standards-free metabolomics—with which they calculate or predict information about multiple properties for molecules of interest in order to generate comprehensive reference libraries and then match experimental data containing the same properties to these libraries, enabling compound identification.
Using this new approach, researchers send chemical structures through machine learning or quantum chemistry programs to accurately predict the experimental properties of the metabolites.
"If we're accurate enough on these predictions then we would theoretically never need to analyze a pure compound again," said Metz. "This collection of tools will shift the current paradigm in metabolomics, and in the near future there are going to be some really good applications to show the research community the benefits of this new approach."

By not having to rely on data from analyses of pure standards to identify small molecules, the standards-free approach allows for the identification of up to 90 percent more chemicals in samples and makes these computational tools highly useful in several application areas, including new drug discovery, chemical forensics, and environmental and biomedical research.
"For example, in new drug design a user would be able to say, 'I've got a certain number of properties with these certain drugs, but they happen to be toxic. Can we predict a compound that would have similar properties but might not be toxic?'" said Metz. "If the right training data could be given to the DarkChem program, DarkChem could then perform that prediction."
Customizable suite of programs
The new approach to standards-free metabolomics identification uses four key tools to generate comprehensive, in silico-derived metabolite reference libraries, and to extract and match experimental data to yield compound identifications:
- In Silico Chemical Library Engine (ISiCLE), a high-performance-computing-friendly, quantum chemistry approach for generating predicted chemical properties.
- DarkChem, a variational autoencoder that learns a continuous numerical or latent representation of molecular structure, which can characterize and expand reference libraries.
- Data Extraction for Integrated Multidimensional Spectrometry (DEIMoS), a modular software tool that can extract features from data collected on multidimensional analytical platforms.
- Multi Attribute Matching Engine (MAME), which matches experimental data to reference libraries based on various chemical attributes.
The tools have been designed to work together, but they can also be used separately. Researchers can customize the different applications based on a client's needs or research areas, creating a completely modular approach.
Advancing a research field
Right now, in the metabolomics community, all researchers identify the same set of molecules in every sample. The reason for that is that they all have the same pure compounds that they purchased to build out their reference libraries.
"Our vision is that by using the standards-free approach you will never be limited by the expanse of small molecules that can be identified in a sample," said Metz. "That's really a game changer for metabolomics. And it's very exciting to see what the next year or so has in store for this."
Provided by Pacific Northwest National Laboratory