Researchers discover molecule's unusual cell-killing mechanism

Soon after Jake Eaton joined the lab of Stuart Schreiber at the Broad Institute of MIT and Harvard in 2015, the postdoctoral researcher became intrigued by some wild theories being debated by his colleagues. The theories centered on a strange small molecule known as ML210, which can kill cells by turning on a cellular process called ferroptosis. Vasanthi Viswanathan, a postdoctoral fellow in the Schreiber lab, had discovered that inducing ferroptosis could kill some drug-resistant cancer cells. Eaton, Viswanathan, and Schreiber believed that understanding how ML210 triggers that process could unlock secrets for how to develop drugs for treating cancers resistant to existing therapies, or prevent resistance in the first place.
Eaton and his colleagues knew that the ML210 compound induces ferroptosis by inhibiting GPX4, a protein that protects cells from oxidative stress. They speculated that it did so by forming a specific type of chemical link, called a covalent bond, with the protein. The problem was, the chemical structure of ML210 showed no obvious ways for it to form covalent bonds, leading to some far-fetched speculation by the skeptical scientists about how it was able to bind GPX4.
"We wondered, 'Is it binding GPX4 in a different way? Is it acting on other nodes in the GPX4 pathway, or doing something entirely new?'," said Eaton. "It was a real chemical mystery."
Now, after years of work, scientists at the Broad Institute, in collaboration with scientists at Bayer, have solved this chemical conundrum. In a study in Nature Chemical Biology, they show that ML210 transforms within the cell into a new molecule, which transforms into a third molecule that then covalently binds GPX4. The mechanism they revealed is extremely unusual and demonstrates an unappreciated way that "pro-drugs" like ML210 can be converted into molecules capable of covalently binding target proteins within cells.
In the work, the scientists describe a new set of compounds that scientists can use to learn more about how cells undergo ferroptosis, a process discovered only a decade ago. In addition, the molecules are starting points for developing compounds that can inhibit GPX4 and kill drug-resistant cancer cells not just in a dish, but potentially also in animal models and even patients.
"I remember saying, 'I fear I'm going to take this ML210 mystery to my grave'," said co-senior author Schreiber, who is a co-founder of and core institute member at the Broad and the Morris Loeb Professor in the Department of Chemistry and Chemical Biology at Harvard University. "Like peeling the many layers of an onion, Jake has revealed, step by step, the fascinating sequence of chemical reactions that cells confer on ML210, which has very little precedence in organic chemistry. It was a brilliant job of molecular sleuthing by Jake."
Detective work
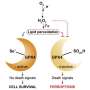
Ferroptosis was first formally described nearly a decade ago by Brent Stockwell, a former student in Schreiber's lab who is now a professor at Columbia University. In ferroptosis, lipid molecules in the cell membrane are oxidized, leading to a buildup of toxic molecules called lipid peroxides that eventually kills the cell. GPX4 protects cells from this kind of death by converting lipid peroxides into nontoxic compounds.
Viswanathan, a transplant from the Stockwell lab into the Schreiber lab, reasoned that a molecule that disrupts GPX4 could be the inspiration for a new kind of cancer drug. However, GPX4 is a challenging target, because its flat, featureless chemical structure has no obvious place to which drugs can bind.
In 2010, a small molecule discovery effort called the Molecular Libraries Probe Production Centers Network, based in part at the Broad Institute, had uncovered a few compounds that bind GPX4 and induce ferroptosis—two of these belong to a class of molecules known as chloroacetamides, which covalently bind GPX4. But these compounds are not good candidates for drug development or in animal studies because they are highly reactive and will disrupt many other proteins, leading to unintended side effects. They are also unlikely to persist in the body long enough to bind and block GPX4.
A third compound from the 2010 screen was ML210, an "outlier compound" that is chemically distinct from the chloroacetamides. Eaton and Viswanathan examined data on ML210 from the Cancer Therapeutics Response Portal, a database developed by researchers at the Broad and sponsored in part by the National Cancer Institute. From the data, it looked like ML210 was acting similarly to the chloroacetamides by forming a covalent bond with GPX4. What stumped the researchers was that ML210 doesn't contain a "covalent warhead," a key chemical structure that would allow it to pair with GPX4 in this way.
Binding mystery
To probe how ML210 inhibits GPX4, the team needed better methods for studying GPX4 in the lab. With colleagues at Bayer, they developed a system to overexpress the protein in mammalian cells and produced assays to study how molecules interact with it.
"One of the great things about this particular industry-academic collaboration has been overcoming some of the problems we've faced in this research," said Eaton. "I don't think we would have overcome those challenges on our own."
The assays confirmed that ML210 was, in fact, inhibiting GPX4 through covalent binding, and that it did so much more precisely than the two chloroacetamides.
This hint led Eaton to further chemical detective work, which revealed that ML210 undergoes a couple of unusual chemical transformations in the cell to gain the ability to bind and inhibit GPX4. ML210 is first transformed into a compound the team dubbed JKE-1674. The cell converts this compound into another unusual molecule called JKE-1777, which is capable of covalently binding GPX4.
Although JKE-1777 is unstable outside of the cell, JKE-1674 and related compounds that the team synthesized are stable and selective for GPX4, and are more suitable than ML210 for use in animal models or perhaps even patients.
Unprecedented machines
In a related paper in the Journal of the American Chemical Society, the researchers describe another set of compounds known as diacylfuroxans that also inhibit GPX4 covalently. While those compounds aren't as selective as ML210 or JKE-1674 and are unlikely to be useful therapeutically, the companion study helped the scientists make sense of the novel observations made in the ML210 study.
"These compounds are molecular machines that are unprecedented in the history of chemical biology for the unusual multi-layered chemical features that underlie their specificity," said co-senior author Vasanthi Viswanathan, a postdoctoral associate in the Schreiber lab.
More work remains to determine what cellular processes guide the transformation of ML210 into its active form, and whether any of the molecules can be used in animal models or even in humans as therapeutic compounds.
If the molecules or variants of them prove promising as new therapeutics, they might give rise to a new class of drugs that could one day help fight drug-resistant tumors.
More information: John K. Eaton et al. Selective covalent targeting of GPX4 using masked nitrile-oxide electrophiles, Nature Chemical Biology (2020). DOI: 10.1038/s41589-020-0501-5
Journal information: Nature Chemical Biology , Journal of the American Chemical Society
Provided by Broad Institute of MIT and Harvard