New approach could accelerate efforts to catalogue vast numbers of cells

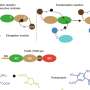
Artistic sketches can be used to capture details of a scene in a simpler image. MIT researchers are now bringing that concept to computational biology, with a novel method that extracts comprehensive samples—called "sketches"—of massive cell datasets that are easier to analyze for biological and medical studies.
Recent years have seen an explosion in profiling single cells from a diverse range of human tissue and organs—such as a neurons, muscles, and immune cells—to gain insight into human health and treating disease. The largest datasets contain anywhere from around 100,000 to 2 million cells, and growing. The long-term goal of the Human Cell Atlas, for instance, is to profile about 10 billion cells. Each cell itself contains tons of data on RNA expression, which can provide insight about cell behavior and disease progression.
With enough computation power, biologists can analyze full datasets, but it takes hours or days. Without those resources, it's impractical. Sampling methods can be used to extract small subsets of the cells for faster, more efficient analysis, but they don't scale well to large datasets and often miss less abundant cell types.
In a paper being presented next week at the Research in Computational Molecular Biology conference, the MIT researchers describe a method that captures a fully comprehensive "sketch" of an entire dataset that can be shared and merged easily with other datasets. Instead of sampling cells with equal probability, it evenly samples cells from across the diverse cell types present in the dataset.
"These are like sketches on paper, where an artist will try to preserve all the important features of a main image," says Bonnie Berger, the Simons Professor of Mathematics at MIT, a professor of electrical engineering and computer science, and head of the Computation and Biology group.
In experiments, the method generated sketches from datasets of millions of cells in a few minutes—as opposed to a few hours—that had far more equal representation of rare cells from across the datasets. The sketches even captured, in one instance, a rare subset of inflammatory macrophages that other methods missed.
"Most biologists analyzing single-cell data are just working on their laptops," says Brian Hie, a Ph.D. student in the Computer Science and Artificial Intelligence Laboratory (CSAIL) and a researcher in the Computation and Biology group. "Sketching gives a compact summary of a very large dataset that tries to preserve as much biological information as possible … so people don't need to use so much computational power."
Joining Hie and Berger on the paper are: CSAIL Ph.D. student Hyunghoon Cho; Benjamin DeMeo, a graduate student at MIT and Harvard Medical School; and Bryan Bryson, an MIT assistant professor of biological engineering.
 and sketches (right) of datasets, where each cell type is a different color. The sketches captured a far more representative sample of the datasets. Credit: Massachusetts Institute of Technology")
Plaid coverings
Humans have hundreds of categories and subcategories of cells, and each cell expresses a diverse set of genes. Techniques such as RNA sequencing capture all cell information in massive tables, where each row represents a cell and each column represents some measurement of gene expression. Cells are points scattered around a sprawling multidimensional space where each dimension corresponds to the expression of a different gene.
As it happens, cell types with similar gene diversity—both common and rare—form similar-sized clusters that take up roughly the same space. But the density of cells within those clusters varies greatly: 1,000 cells may reside in a common cluster, while the equally diverse rare cluster will contain 10 cells. That's a problem for traditional sampling methods that extract a target-size sample of single cells.
"If you take a 10-percent sample, and there are 10 cells in a rare cluster and 1,000 cells in a common cluster, you're more likely to grab tons of common cells, but miss all rare cells," Hie says. "But rare cells can lead to important biological discoveries."
The researchers modified a class of algorithm that lays shapes over datasets. Their algorithm covers the entire computational space with what they call a "plaid covering," which is like a grid of equal-sized squares but in many dimensions. It only lays these multidimensional squares where there's at least one cell, and skips over any empty regions. In the end, the grid's empty columns will be much wider or skinnier than occupied columns—hence the "plaid" description. That technique saves tons of computation to help the covering scale to massive datasets.
Capturing rare cells
Occupied squares may contain only one cell or 1,000 cells, but they will all have the exact same sampling weight. The algorithm then finds a target sample—of, say, 20,000 cells—by selecting a set number of cells from each occupied square uniformly, at random. The resulting sketch contains a far more equal distribution of cell types—for example, 10 common cells from a cluster of 100 and eight rare cells from a cluster of 10.
"We take advantage of these cell types occupying similar volumes of space," Hie says. "Because we sample according to volume, instead of density, we get a more even coverage of the biological space … and we're naturally preserving the rare cell types."
They applied their sketching method to a dataset of around 250,000 umbilical cord cells that contained two subsets of a rare macrophages—inflammatory and anti-inflammatory. All other traditional sampling methods clustered both subsets together, while the sketching method separated them. Additional in-depth studies of these macrophage subpopulations could help reveal insight into inflammation and how to modulate inflammatory processes in response to disease, the researchers say.
"That's a benefit in working at the interface of fields," Berger says. "We're trained as mathematicians, but we understand what biological data science problems are, so we can bring the best technologies to their analysis."
More information: Hie et al. Geometric Sketching Compactly Summarizes the Single-Cell Transcriptomic Landscape: Supplementary Information, BioRxiv (2019). DOI: 10.1101/536730
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.