Researchers provide new method to boost clean energy research
, the Allan and Myra Ferguson Chair of Chemical Engineering at UD, was co-author on the paper published in the journal Nature Chemistry. Credit: University of Delaware")
Electrochemical energy systems—processes by which electrical energy is converted to chemical energy—are at the heart of establishing more efficient generation and storage of intermittent energy from renewable sources in fuel cells and batteries.
The powerhouse substances known as catalysts, which are used to accelerate chemical reactions, are key players in these systems. The size and efficiency of fuel cells, for example, could greatly benefit from using high-performance catalysts.
Producing better catalysts is easier said than done, however. A catalyst's usefulness is partially based on the amount and quality of its active sites, due to the sites' specific geometry and electronic properties. Engineering these sites can be an arduous, inefficient process.
Now, researchers at the University of Delaware have revolutionized the way in which scientists can design catalyst structures. Their work, featured in the latest issue of leading science journal Nature Chemistry, has established a new approach for managing highly structure-sensitive chemistries to achieve the highest possible activity while considering catalyst stability.
"Optimizing catalysts at the atomic level has been a long-standing problem, as the active centers are typically unknown, and how to best pack them together to perform the chemistry has remained elusive," said Dion Vlachos, Allan and Myra Ferguson Chair of Chemical Engineering at UD and co-author on the paper. "As we engineer materials for improved performance, the stability of materials is critical. Our method is the first to address both crystal engineering with atomic precision and material stability."
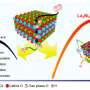
According to the researchers, what sets their method apart is the streamlining of the material synthesis, using computers to create microscopic variations—or nanodefects—on a catalyst's surface.
"In the past, researchers have modeled different active sites one at a time, which is very time-consuming," says co-author Marcel Nunez, who earned his doctorate in chemical and biomolecular engineering at UD and now serves as a design engineer at Intel. "Our approach is automated. It's really the first of its kind, helping to make catalysts easier to synthesize and more stable during chemical reactions."
Josh Lansford, a doctoral candidate in the Vlachos lab and also a co-author on the paper, emphasized that, while the computations start on a small scale—quantum, in this case—the results are anything but.
"It's all about restructuring the surface of the catalyst to decrease the energy necessary to make the reaction go," he said. "The more active the site, the higher the electric current, which leads to a faster reaction and more powerful fuel cell."
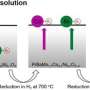
The researchers demonstrated the effectiveness of their new methodology using a process called the oxygen reduction reaction (ORR), which is often used to generate power in fuel cells for transportation. Because oxygen is plentiful in the earth's atmosphere, ORR is an ideal method for producing portable power sources that don't emit carbon dioxide (CO2).
While fuel cells have yet to be economically viable on a large scale, the authors said they hope their breakthrough will help change that, opening new avenues for cleaner and more economical energy production.
"The long-term vision for our methodology is that it will be used to design the desired catalyst structure on computers," Nunez said. "The catalyst would then be synthesized and characterized in the laboratory and used in fuel cells, having a higher performance than the current industrial standard. Our approach edges us towards the economic feasibility of clean fuel cell vehicles."
More information: M. Núñez et al. Optimization of the facet structure of transition-metal catalysts applied to the oxygen reduction reaction, Nature Chemistry (2019). DOI: 10.1038/s41557-019-0247-4
Journal information: Nature Chemistry
Provided by University of Delaware