Anti-evolvability drugs could slow antibiotic resistance in bacteria
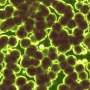
 in living E. coli challenged with sub-lethal ciprofloxacin. Credit: John P Pribis")
The failure of existing antibiotics to combat infections is a major health threat worldwide. While the traditional strategy for tackling drug resistance has been to develop new antibiotics, a more sustainable long-term approach may be preventing bacteria from evolving it in the first place. Until now, one major hurdle to this approach is that it has not been clear how antibiotics induce new mutations.
In a study publishing April 1 in the journal Molecular Cell, researchers found that one mechanism by which antibiotics induce drug-resistance mutations in bacteria is by triggering the generation of high levels of toxic molecules called reactive oxygen species (ROS). Additionally, treatment with a ROS-reducing drug approved by the U.S. Food and Drug Administration for other purposes prevented these antibiotic-induced mutations. However, future preclinical trials are needed to assess the effectiveness of such drugs in combatting resistance evolution and promoting the clearance of infections in animal models.
"We wanted to understand the molecular mechanism underlying the evolutionary arms race that pathogenic bacteria wage against our immune systems, and against antibiotics," says senior study author Susan Rosenberg of Baylor College of Medicine. "This is motivated by the hope of being able to make or identify a fundamentally new kind of drug to slow bacterial evolution. Not an antibiotic, which kills cells or stops their proliferation, but an anti-evolvability drug, which would slow evolution, allowing our immune systems and drugs to defeat infections."
To understand how antibiotics induce new mutations, first author John Pribis of Baylor College of Medicine, Rosenberg, and their team began by exposing Escherichia coli to low doses of the antibiotic ciprofloxacin, which induces DNA breaks. Approximately 10%-25% of the cell population generated high levels of ROS, which transiently activated a pronounced stress response.
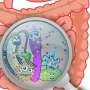
, which induce the general stress response (red), and an intermediate state with both high ROS and stress-response activity in the same cells (orange). Credit: John P Pribis")
But surprisingly, this stress response allowed the "gambler" subpopulation to switch repair of the DNA breaks from accurate to error-prone, resulting in new mutations that promoted resistance to antibiotics that had never before been encountered. According to the authors, the development of a transient gambler subpopulation may be a bet-hedging strategy that could drive the evolution of resistance to new antibiotics without risk to most cells.
"This particular mechanism is likely to be important for resistance to quinolones—very widely used antibiotics for which clinical resistance is common and occurs by new mutations in the clinic," Rosenberg says. "It is likely also to illuminate formation of resistance to other antibiotics, in which the main route to resistance is new mutations, as opposed to those antibiotics for which the main route is acquisition of resistance genes from other bacteria."
In additional experiments, the researchers found that exposure to the ROS-reducing drug edaravone, which is approved for the treatment of stroke and amyotrophic lateral sclerosis, effectively inhibited the stress response and ciprofloxacin-induced mutations without altering antibiotic activity. "These data serve as a proof-of-concept for small-molecule inhibitors that could be administered with antibiotics to reduce resistance evolution by impeding differentiation of gamblers, without harming antibiotic activity," Rosenberg says. "Edaravone is approved for human use, so if it proves useful in preclinical trials, it could be fast-tracked for human trials, because it has a known safety profile."
"Drugs like this could be used with standard antibiotics to slow evolution of resistance," she says. "These could potentially extend the use of current antibiotics, and possibly work as mono-therapies by tilting the evolutionary battle in favor of the immune system."
In future studies, Rosenberg and her team will test whether anti-evolvability drugs prevent antibiotic resistance and improve clinical outcomes in animals infected with pathogenic bacteria. They also plan to look for additional drug targets. "This is not the sole molecular mechanism of stress-induced mutagenesis," Rosenberg says. "We wish to discover others that could be similarly impactful in understanding and combatting resistance evolution."
More information: Molecular Cell, Pribis et al.: "Gamblers: an Antibiotic-induced Evolvable Cell Subpopulation Differentiated by Reactive-oxygen-induced General Stress Response" www.cell.com/molecular-cell/fu … 1097-2765(19)30148-0 , DOI: 10.1016/j.molcel.2019.02.037
Journal information: Molecular Cell
Provided by Cell Press