Atomic-scale simulation of antiarrhythmic drug interaction with cardiac cells

To unravel the mysterious mechanisms of drug potency for the treatment of cardiac arrhythmias, a group of researchers at UC Davis have developed novel simulations that provide insights on vital atomic-scale drug-cardiac cells interactions.
These simulations, published today in PNAS (Proceedings of the National Academy of Sciences), may lead the way to better development of new antiarrhythmic drugs targeting voltage-gated sodium (NaV) channels, specialized protein molecules in the cardiac cell membrane.
Sodium channels serve as gatekeepers regulating the electric activity of cardiac cells. When the electric signals coordinating the heartbeats are not working properly, the heart may experience irregular heartbeats and is considered in an arrhythmic state.
A class of antiarrhythmic drugs works on NaV channels to influence the heart's electrical activity and its beat. Yet, the longstanding failures in drug treatment of heart rhythm disturbances stem mainly from the inability to predict the impact of developed drugs on the activity of NaV and other cardiac ion channels.
"Before our study, there has been no effective preclinical methodology to differentiate useful or potentially harmful drugs at the molecular level," said Vladimir Yarov-Yarovoy, associate professor at the UC Davis Department of Physiology and Membrane Biology.
"To develop and screen novel drugs for treatment of cardiovascular diseases and to minimize their side effects, there is a need to understand the mechanism of antiarrhythmic drug interactions with NaV channels at an atomic scale," he said.
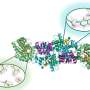
Thanks to several technological breakthroughs and an increasing number of available high-resolution structures of ion channels, such as NaV, researchers are now able to simulate these structures and to modulate the activity of the heart cells by studying their interactions at atomic resolution. The researchers were able to build a model of the human NaV channel based on the closely resembling structure of the electric eel NaV channel using Rosetta computational modeling software.
NaV channels open to allow the sodium ions to flow into the cardiac cells and close within milliseconds. When the drug molecules enter these channels, they bind tightly to the receptor site within the protein preventing the sodium ions from entering the cell and blocking the channel conduction. This change in conduction affects the heart's electric activity and its beat.
In the developed atomic model simulations, two drug molecules are seen transiting into the channel central pore and binding to the receptor site of the protein forming the "hot spots", areas where most favorable drug-protein interaction occur. This binding activity triggers what is known as a high affinity state of the channel.
"High affinity state of the channel is considered the most important state to study drug-protein binding mechanism. Now and for the first time, we can understand how this binding process happens at atomic scale," Yarov-Yarovoy added.
Multi-microsecond simulations of lidocaine (antiarrhythmic and local anesthetic drug) interacting with sodium channels revealed a channel pore access pathway through the intracellular gate and a novel access pathway through a relatively small lateral opening known as fenestration.
Combining molecular modeling software with simulations to study drug-channel interactions is a novel approach that allows future automated virtual drug screening. This technology can be applied to any ion channel and would benefit multiple treatments. Ultimately, this approach advances precision medicine by predicting individual patient responses to drug therapy based on the specific ion channel mutation the patient has.
More information: Phuong T. Nguyen et al, Structural basis for antiarrhythmic drug interactions with the human cardiac sodium channel, Proceedings of the National Academy of Sciences (2019). DOI: 10.1073/pnas.1817446116
Journal information: Proceedings of the National Academy of Sciences
Provided by UC Davis