Fluorescence-activating beta-barrel protein made from scratch for first time

For the first time, scientists have created, entirely from scratch, a protein capable of binding to a small target molecule. Researchers from the University of Washington School of Medicine report the advance in the Sept. 12 issue of the journal Nature.
Previously, such small-molecule binding proteins have been made by altering proteins already existing in nature. That approach significantly limited the possibilities. The ability to make such proteins from scratch, or "de novo," opens the way for scientists to create proteins unlike any found in nature. These proteins can be custom-designed with high precision and affinity to bind to and act on specific small molecule targets.
The lead authors of the paper are Jiayi Dou and Anastassia A. Vorobieva, both senior fellows in the lab of senior author David Baker, professor of biochemistry at the UW School of Medicine and director of the Institute of Protein Design at UW Medicine. Baker is also an investigator at the Howard Hughes Medical Institute.
The technique should have wide application in research, medicine and industry, according to Baker.
"The successful de novo design of custom-built proteins with small-molecule binding activity sets the stage for the creation of increasingly sophisticated binding proteins that will not have the limitations seen with proteins that have been designed by altering existing protein structures," he explained.
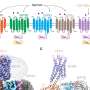
To make the protein, the researchers had to achieve another first: Creating from scratch a cylinder-shaped protein called a beta-barrel. The beta-barrel structure was ideal because one end of the cylinder could be designed to stabilize the protein, while the other end could be used to create a cavity that can serve as the binding site for the target molecule.
Proteins are made of long chains of amino acids. Once synthesized, these chains fold into precise shapes that allow the proteins to perform their functions. The shapes these chains assume are typically incredibly convoluted, but two regular features often occur: alpha-helices, which form when the sections chain winds around a central axis, and sheet-like structures, called beta-sheets.
Beta-sheets form when two or more sections from different parts of the amino acid chain, because of folding, run side-by-side in 3-D space. These sections are "stitched together" by hydrogen bonds, creating a sheet-like structure. These beta-sheets, in turn, can assemble into barrel-like structures, called beta-barrels. In nature, beta-barrels proteins bind a wide range of small molecules.
To design the new protein, Dou and Vorobieva used a software platform, developed in the Baker lab, called Rosetta. It can predict what shape a particular chain of amino acids will assume after synthesis and can tell how changing individual amino acids along the chain may alter that shape. This predictive power makes it possible to test out different combinations of amino acids to design a protein with the desired shape and function.
To create the cavity, the researchers used a powerful new docking algorithm, called "Rotamer Interaction Field" (RIF), developed by William Sheffler, a senior research scientist in the Baker lab. RIF rapidly identifies all potential structures of cavities that fulfill the pre-requisites for binding specific molecules.
Equipped with the new RIF docking methods, Dou, Vorobieva and Sheffler designed the beta barrels to bind a compound called DFHBI, a component similar to what is housed inside green fluorescent protein, which fluoresces when exposed to certain frequencies of light. Green fluorescent protein is routinely used in biological research to locate molecules and structures within living organism and to track their movement.

In their paper, the researcher demonstrate that their custom-designed protein avidly bound and activated the DFHBI compound.
"It worked in bacterial, yeast and mammalian cells," said Dou, "and being half the size of green fluorescent protein should be very useful to researchers."
Baker said that the approach will allow researchers to explore an effectively unlimited set of backbone structures with shapes customized to bind the molecule of interest.
"Equally important," he added, "it greatly advances our understanding of the determinants of protein folding and binding beyond what we have learned from describing existing protein structures."
More information: Jiayi Dou et al, De novo design of a fluorescence-activating β-barrel, Nature (2018). DOI: 10.1038/s41586-018-0509-0
Journal information: Nature
Provided by University of Washington