Automating molecule design to speed up drug development

Designing new molecules for pharmaceuticals is primarily a manual, time-consuming process that's prone to error. But MIT researchers have now taken a step toward fully automating the design process, which could drastically speed things up—and produce better results.
Drug discovery relies on lead optimization. In this process, chemists select a target ("lead") molecule with known potential to combat a specific disease, then tweak its chemical properties for higher potency and other factors.
Often, chemists use expert knowledge and conduct manual tweaking of molecules, adding and subtracting functional groups—atoms and bonds responsible for specific chemical reactions—one by one. Even if they use systems that predict optimal chemical properties, chemists still need to do each modification step themselves. This can take hours for each iteration and may still not produce a valid drug candidate.
Researchers from MIT's Computer Science and Artificial Intelligence Laboratory (CSAIL) and Department of Electrical Engineering and Computer Science (EECS) have developed a model that better selects lead molecule candidates based on desired properties. It also modifies the molecular structure needed to achieve a higher potency, while ensuring the molecule is still chemically valid.
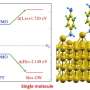
The model basically takes as input molecular structure data and directly creates molecular graphs—detailed representations of a molecular structure, with nodes representing atoms and edges representing bonds. It breaks those graphs down into smaller clusters of valid functional groups that it uses as "building blocks" that help it more accurately reconstruct and better modify molecules.
"The motivation behind this was to replace the inefficient human modification process of designing molecules with automated iteration and assure the validity of the molecules we generate," says Wengong Jin, a Ph.D. student in CSAIL and lead author of a paper describing the model that's being presented at the 2018 International Conference on Machine Learning in July.
Joining Jin on the paper are Regina Barzilay, the Delta Electronics Professor at CSAIL and EECS and Tommi S. Jaakkola, the Thomas Siebel Professor of Electrical Engineering and Computer Science in CSAIL, EECS, and at the Institute for Data, Systems, and Society.
The research was conducted as part of the Machine Learning for Pharmaceutical Discovery and Synthesis Consortium between MIT and eight pharmaceutical companies, announced in May. The consortium identified lead optimization as one key challenge in drug discovery.
"Today, it's really a craft, which requires a lot of skilled chemists to succeed, and that's what we want to improve," Barzilay says. "The next step is to take this technology from academia to use on real pharmaceutical design cases, and demonstrate that it can assist human chemists in doing their work, which can be challenging."
"Automating the process also presents new machine-learning challenges," Jaakkola says. "Learning to relate, modify, and generate molecular graphs drives new technical ideas and methods."
Generating molecular graphs
Systems that attempt to automate molecule design have cropped up in recent years, but their problem is validity. Those systems, Jin says, often generate molecules that are invalid under chemical rules, and they fails to produce molecules with optimal properties. This essentially makes full automation of molecule design infeasible.
These systems run on linear notations of molecules, called "simplified molecular-input line-entry systems," or SMILES, where long strings of letters, numbers, and symbols represent individual atoms or bonds that can be interpreted by computer software. As the system modifies a lead molecule, it expands its string representation symbol by symbol—atom by atom, and bond by bond—until it generates a final SMILES string with higher potency of a desired property. In the end, the system may produce a final SMILES string that seems valid under SMILES grammar, but is actually invalid.
The researchers solve this issue by building a model that runs directly on molecular graphs, instead of SMILES strings, which can be modified more efficiently and accurately.
Powering the model is a custom variational autoencoder—a neural network that "encodes" an input molecule into a vector, which is basically a storage space for the molecule's structural data, and then "decodes" that vector to a graph that matches the input molecule.
At encoding phase, the model breaks down each molecular graph into clusters, or "subgraphs," each of which represents a specific building block. Such clusters are automatically constructed by a common machine-learning concept, called tree decomposition, where a complex graph is mapped into a tree structure of clusters—"which gives a scaffold of the original graph," Jin says.
Both scaffold tree structure and molecular graph structure are encoded into their own vectors, where molecules are group together by similarity. This makes finding and modifying molecules an easier task.
At decoding phase, the model reconstructs the molecular graph in a "coarse-to-fine" manner—gradually increasing resolution of a low-resolution image to create a more refined version. It first generates the tree-structured scaffold, and then assembles the associated clusters (nodes in the tree) together into a coherent molecular graph. This ensures the reconstructed molecular graph is an exact replication of the original structure.
For lead optimization, the model can then modify lead molecules based on a desired property. It does so with aid of a prediction algorithm that scores each molecule with a potency value of that property. In the paper, for instance, the researchers sought molecules with a combination of two properties—high solubility and synthetic accessibility.
Given a desired property, the model optimizes a lead molecule by using the prediction algorithm to modify its vector—and, therefore, structure—by editing the molecule's functional groups to achieve a higher potency score. It repeats this step for multiple iterations, until it finds the highest predicted potency score. Then, the model finally decodes a new molecule from the updated vector, with modified structure, by compiling all the corresponding clusters.
Valid and more potent
The researchers trained their model on 250,000 molecular graphs from the ZINC database, a collection of 3-D molecular structures available for public use. They tested the model on tasks to generate valid molecules, find the best lead molecules, and design novel molecules with increase potencies.
In the first test, the researchers' model generated 100 percent chemically valid molecules from a sample distribution, compared to SMILES models that generated 43 percent valid molecules from the same distribution.
The second test involved two tasks. First, the model searched the entire collection of molecules to find the best lead molecule for the desired properties—solubility and synthetic accessibility. In that task, the model found a lead molecule with a 30 percent higher potency than traditional systems. The second task involved modifying 800 molecules for higher potency, but are structurally similar to the lead molecule. In doing so, the model created new molecules, closely resembling the lead's structure, averaging a more than 80 percent improvement in potency.
The researchers next aim to test the model on more properties, beyond solubility, which are more therapeutically relevant. That, however, requires more data. "Pharmaceutical companies are more interested in properties that fight against biological targets, but they have less data on those. A challenge is developing a model that can work with a limited amount of training data," Jin says.
Provided by Massachusetts Institute of Technology
This story is republished courtesy of MIT News (web.mit.edu/newsoffice/), a popular site that covers news about MIT research, innovation and teaching.