New statistical method for evaluating reproducibility in studies of genome organization
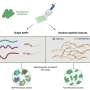
 is first smoothed in order to allow researchers to see trends in the data more clearly. Step 2: The data is stratified based on distance to account for the overabundance of nearby interactions in Hi-C data. Credit: Li Laboratory, Penn State University")
A new statistical method to evaluate the reproducibility of data from Hi-C—a cutting-edge tool for studying how the genome works in three dimensions inside of a cell—will help ensure that the data in these "big data" studies is reliable.
"Hi-C captures the physical interactions among different regions of the genome," said Qunhua Li, assistant professor of statistics at Penn State and lead author of the paper. "These interactions play a role in determining what makes a muscle cell a muscle cell instead of a nerve or cancer cell. However, standard measures to assess data reproducibility often cannot tell if two samples come from the same cell type or from completely unrelated cell types. This makes it difficult to judge if the data is reproducible. We have developed a novel method to accurately evaluate the reproducibility of Hi-C data, which will allow researchers to more confidently interpret the biology from the data."
The new method, called HiCRep, developed by a team of researchers at Penn State and the University of Washington, is the first to account for a unique feature of Hi-C data—interactions between regions of the genome that are close together are far more likely to happen by chance and therefore create spurious, or false, similarity between unrelated samples. A paper describing the new method appears in the journal Genome Research.
"With the massive amount of data that is being produced in whole-genome studies, it is vital to ensure the quality of the data," said Li. "With high-throughput technologies like Hi-C, we are in a position to gain new insight into how the genome works inside of a cell, but only if the data is reliable and reproducible."
Inside the nucleus of a cell there is a massive amount of genetic material in the form of chromosomes—extremely long molecules made of DNA and proteins. The chromosomes, which contain genes and the regulatory DNA sequences that control when and where the genes are used, are organized and packaged into a structure called chromatin. The cell's fate, whether it becomes a muscle or nerve cell, for example, depends, at least in part, on which parts of the chromatin structure is accessible for genes to be expressed, which parts are closed, and how these regions interact. HiC identifies these interactions by locking the interacting regions of the genome together, isolating them, and then sequencing them to find out where they came from in the genome.

"It's kind of like a giant bowl of spaghetti in which every place the noodles touch could be a biologically important interaction," said Li. "Hi-C finds all of these interactions, but the vast majority of them occur between regions of the genome that are very close to each other on the chromosomes and do not have specific biological functions. A consequence of this is that the strength of signals heavily depends on the distance between the interaction regions. This makes it extremely difficult for commonly-used reproducibility measures, such as correlation coefficients, to differentiate Hi-C data because this pattern can look very similar even between very different cell types. Our new method takes this feature of Hi-C into account and allows us to reliably distinguish different cell types."
"This reteaches us a basic statistical lesson that is often overlooked in the field," said Li. "Quite often, correlation is treated as a proxy of reproducibility in many scientific disciplines, but they actually are not the same thing. Correlation is about how strongly two objects are related. Two irrelevant objects can have high correlation by being related to a common factor. This is the case here. Distance is the hidden common factor in the Hi-C data that drives the correlation, making the correlation fail to reflect the information of interest. Ironically, while this phenomenon, known as the confounding effect in statistical terms, is discussed in every elementary statistics course, it is still quite striking to see how often it is overlooked in practice, even among well-trained scientists."
The researchers designed HiCRep to systematically account for this distance-dependent feature of Hi-C data. In order to accomplish this, the researchers first smooth the data to allow them to see trends in the data more clearly. They then developed a new measure of similarity that is able to more easily distinguish data from different cell types by stratifying the interactions based on the distance between the two regions. "This is like studying the effect of drug treatment for a population with very different ages. Stratifying by age helps us focus on the drug effect. For our case, stratifying by distance helps us focus on the true relationship between samples."
To test their method, the research team evaluated Hi-C data from several different cell types using HiCRep and two traditional methods. Where the traditional methods were tripped up by spurious correlations based on the excess of nearby interactions, HiCRep was able to reliably differentiate the cell types. Additionally, HiCRep could quantify the amount of difference between cell types and accurately reconstruct which cells were more closely related to one another.
Journal information: Genome Research
Provided by Pennsylvania State University