New digital method enhances understanding of changes in DNA's makeup

Scientists have developed a computational method to detect chemical changes in DNA that highlight cell diversity and may lead to a better understanding of cancer.
In the European study, published in Nucleic Acids Research and involving Newcastle University, experts have established a bioinformatics method that can be applied in hematopoiesis, the process of blood formation from stem cells.
The information of how to form the different cells in our body is encoded in our genome, which is composed by the four letters of DNA.
Each cell type is characterised by its distinct epigenome, a makeup of the genome composed of many different proteins that leave "open" or "closed" different parts of the genome.
Using different makeups, the same genome in one person can encode the necessary information to develop all the different tissues and organs.
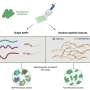
The new method developed allows the automatic analysis of multiple epigenomes to identify the genomic locations where the necessary makeup changes to form both healthy and diseased cell types.
Disease biomarkers
Dr Daniel Rico, Research Fellow at Newcastle University's Institute of Cellular Medicine, said: "We have the technology to reveal the different epigenomic makeups and this is generating a significant amount of data.
"We can seriously talk about "big data" in epigenomics research. However, the main bottleneck is to translate all this data into useful information to get insight into biological mechanisms.
"The new method that we have developed will allow researchers to identify the key regions in the genome that show differential makeups depending of the cell types.
"As many diseases are associated to disease-specific epigenomic makeups, this method will be particularly useful to identify the key regions in the genome where the makeup deviates from the healthy state.
"This will allow the development of new disease biomarkers and, hopefully, open a new path for developing therapies targeting the epigenomes."
The method allows the integration of a variety of epigenomic datasets to classify different samples and automatically identify genomic regions in which changes affect the definition of cell type.
Genome makeup
The human genome is a dull sequence of letters but it becomes alive thanks to the help of the epigenome.
The genome is like a book in which letters follow each other without empty spaces, full stops or commas. It would be very hard to read this book, utterly impossible to understand it.
However, with the simple addition of punctuation marks we can read and understand the meaning of that apparently dull sequence of letters.
This is the great task accomplished by the epigenome, which is composed of chemical changes on the DNA that allow us and the cell to understand how to read and interpret the genome.
For this reason, studying the epigenome is important to understanding how development can give rise to the large variety of cell types forming tissues and organs, all starting from a single cell and a single genome.
The epigenome is also often involved in explaining how a healthy cell gives rise to a tumour after a malignant transformation. But, despite the collections of data available until now, locating those regions of the genome with chemical changes and type of changes remains a challenge.
These chemical changes are produced during development and throughout life as an effect of external factors, such as lifestyle, and they can be triggers for diseases like cancer. Identifying epigenetic biomarkers is fundamental to enable new diagnostic and treatment options.
Molecular classifications have been widely used taking into account gene expression levels as biomarkers, using their on/off state.
This method lets researchers identify those regions that are involved in regulating the on/off gene state like biological switches. These regions could be used as epigenetic biomarkers that complement the actual molecular classifications.
Important development
Enrique Carrillo-de-Santa-Pau, co-first author of the study, from the Spanish National Cancer Research Centre, said: "The development of this type of methods is very important.
"Up to now differences between cell types had mostly been characterised at the levels of genes that are either switched on or off, that is the final product of the epigenomic regulation, but we did not know where the switches for these genes were (encoded in the epigenome).
"This acquired knowledge is fundamental to enable new therapies based on acting on the correct switches in cases where the cell loses control in diseases such as cancer.
"Understanding this level of regulation will take us one step further in the personalised medicine agenda."
It is hoped that this method will allow scientists to identify new epigenetic biomarkers, which may be used in personalised medicine diagnosis and treatment.
More information: Automatic identification of informative regions with epigenomic changes associated to hematopoiesis, Nucleic Acids Research, doi.org/10.1093/nar/gkx618
Journal information: Nucleic Acids Research
Provided by Newcastle University