Small scale, big improvements

Methods to improve water purification or build better batteries are problems that have challenged scientists for decades. Advances have inched forward, but rising demand moves the finish line further and further away.
At the same time, the chemical reactions that make these improvements possible occur at scales invisible to the naked eye (the atomic scale) where liquids and solid surfaces meet, making the work even more difficult.
Knowing how these chemical interactions occur at the solid-liquid interface is critical in problems of great interest to the Department of Energy (DOE), particularly as it relates to environmental and water quality issues that may be affected by large-scale energy production activities.
Now, a new technique developed by a team including University of Delaware Prof. Neil Sturchio and colleagues at Argonne National Laboratory and the University of Illinois at Chicago has produced real-time observations documenting the chemical reactions that happen between liquids and solids.
The technique provides data that can be used to improve predictions of how nutrients and contaminants will move in natural systems or to gauge the effectiveness of water purification methods where ion exchange is critical to sanitization.
It also may help scientists tease out limiting factors to supercapacitors—robust energy storage devices that are often used over common batteries to power consumer electronics, hybrid vehicles, even large industrial-scale power.
Energy exchange in chemical reactions
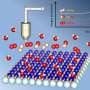
Sturchio, a geochemist, has studied mineral/water interactions for 25 years with funding from DOE. He and his collaborators recently demonstrated a new way of studying the microscopic structure and processes that occur where minerals and water meet, using X-ray beams to trigger the reactions while capturing images of their effects on the mineral surface.
Now using a method called Resonant Anomalous X-Ray Reflectivity (RAXR), the researchers are able to go one step further and distinguish the identity of the element being studied.
"With our previous methods, we could see the atomic-scale electron density profile of the interfacial region—a nanometer-thick zone including the mineral surface and the adjacent solution—but couldn't uniquely identify the atomic layers," said Sturchio, professor and chair of the Department of Geological Sciences in UD's College of Earth, Ocean, and Environment.
The technique requires a high-quality crystal so the researchers selected mica, a mineral similar in structure to the abundant clay minerals in soils that produces an atomically flat crystal useful in laboratory investigations of interfacial properties.
The researchers reflected an intense X-ray beam off a mica sample in alternating contact with two different salt solutions containing rubidium and sodium chloride. By changing the angle of the beam, scientists were able to scan the interfacial profile at atomic scale. By changing the energy of the beam at a fixed angle, they could isolate the distribution of rubidium ions in the interfacial region.
"In this case, we can tune in and ask specifically where is the rubidium? How is it attached to the mica crystal and how is it released to the solution?" he said.
According to Sturchio, most chemical reactions in groundwater and in the atmosphere, as well as during industrial processes including water purification and some forms of energy storage, happen at surfaces such as electrodes or particles. As a chemical reaction occurs, ions are kicked off or pulled on and energy is exchanged. Understanding quantitatively how the ions are exchanged at this scale can be used to design chemical processes to improve water purification or understand how contaminants are transported in soil and groundwater.
In this project, the researchers wanted to see what it would take to get the rubidium, an alkali metal, to release from the mica surface once it was attached. They accomplished this by quickly changing the solution flowing over the mica crystal from rubidium chloride to a more concentrated sodium chloride, then timed the reaction to determine how long it took for the rubidium ions to release (desorb) from the mica and for the sodium chloride ions to take their place (adsorb).
Generally, adsorption reactions are thought to occur in milliseconds, but here it took 25 seconds for the rubidium to release from the surface (desorption) and the sodium ions to take its place (adsorption).
The closer the rubidium was to the mineral/water interface, the more fixed its position became (because of electrostatic energy - the kind that makes a balloon stick to a wall after you rub it against a sweater) and the more energy was required to pry it away from the mica. Conversely, the more water molecules between the crystal's surface and the rubidium ion, the more wiggle room the rubidium had in its position and the less energy it took to break away. The experiments helped to quantify the minute amounts of energy transferred during alkali ion exchange at this interface, and the involvement of water molecules in the reaction mechanism.
The reaction was slower than the researchers anticipated, and while further study is required, they agree the results provide evidence for understanding the timeframes necessary for desired reactions to occur.
By contrast, when the solutions were switched back, the rubidium adsorbed onto the mica surface much more rapidly than it desorbed, by shedding its attached water molecules, demonstrating that hydration is important to the reaction.
"To design an industrial process you need to know exactly what's happening at the surface," Sturchio said. "As far as we know, this is the first time anyone has documented such detailed information on how these ion exchange reactions are happening at a mineral surface in contact with water, and in this case, we have good evidence for how long it actually takes."
More information: Sang Soo Lee et al. Real-time observation of cation exchange kinetics and dynamics at the muscovite-water interface, Nature Communications (2017). DOI: 10.1038/ncomms15826
Journal information: Nature Communications
Provided by University of Delaware