January 11, 2017 report
New research explains hydrophobicity
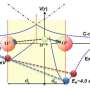
 Frequency of νOD as a function of projection of electric field ED…O. Contributions of the H-bonded pair (yellow) to ED…O are ignored. (B) Distribution of water molecules as a function of distance to atom D and projection of electric field ED…O. Red, blue, green, and yellow systematically correspond to classes I and II, intercalating waters, and the monitored H-bonded water pair, respectively. Class I and II waters are located on the vertices of two fused tetrahedrons. (C) The environment of two H-bonded waters in ice. Waters centered on the vertices of tetrahedrons are marked by red or blue depending on their class. The circle shows the cutoff distance for intercalating waters. (D) The environment of the two H-bonded waters in bulk liquid water. Average electric field vectors and dipole moments for intercalating waters are shown as orange and green arrows, respectively. The resulting ED…O is shown as a red arrow. To accommodate the intercalating water molecule, the angle O-D…O of the H-bonded pair must bend. Only ∼6% of the H-bonded pairs in the bulk water have intercalating waters. (E) The environment of the two H-bonded waters in the first solvation layer of methane. Intercalating water molecules are replaced by methane—the resulting ED…O is larger than that of bulk water (red arrow). Credit: (с) Joze Grdadolnik et al. Origin of hydrophobicity and enhanced water hydrogen bond strength near purely hydrophobic solutes, Proceedings of the National Academy of Sciences (2017). DOI: 10.1073/pnas.1612480114")
(Phys.org)—The hydrophobic effect is a fundamental aspect of biochemical processes. Hydrophilic, or water-loving, solutes tend to be miscible in water, while hydrophobic, or water-fearing, solutes tend to aggregate in such a way as to minimize the number of water-solute interactions.
It is this effect that dictates important processes such as protein folding or intercellular transport. While hydrophobic effects play a key role in life's most fundamental processes, the actual cause of the effect is a topic of debate.
Researchers from the National Institute of Chemistry in Slovenia have, for the first time, experimentally and computationally confirmed the classical view of hydrophobic hydration, which says that hydrogen bonds near a hydrophobic solute become stronger, simulating bonding behavior seen in ice or in clathrates. Their work appears in the Proceedings of the National Academy of Sciences.
"Understanding the fundamental properties of hydrophobicity may bring crucial advances in several important biochemical processes: protein folding, aggregation of protein subunits into complex quaternary structures, molecular recognition (ligand-receptor, enzyme-substrate), and aggregation of amphiphilic lipids into bilayers, micelles, cell membranes, and organelles," Dr. Franc Avbelj, co-author of the paper, told Phys.org.
There are several camps in the hydrophobicity debate: the classical view, the dynamic view, and alternative views. The classical view assumes that in the presence of hydrophobic solute molecules water's hydrogen bonds near the solute (i.e., within the first solvation shell) become enhanced forming what the authors call "transient, semi-ordered clathrate-like clusters." Sometimes these clusters are called "icebergs" indicating patches of water with stronger hydrogen bonds, reminiscent of ice.
The dynamic view differs in that it assumes no change to hydrogen bonding, but rather a change in the molecular rotational motion of water molecules near the solvent. Specifically, the dynamic views' physical background is in the specific jump mechanism of the rotational relaxation of water molecules. Water reorientation relies on bifurcated hydrogen bonds, but hydrophobic solutes hinder the formation of these bonds, thus slowing down the reorientation of water molecules.
Other views say that the hydrophobic effect occurs because solute molecules cannot fit within a cavity between hydrogen bonds in water. Or, others speculate that van der Waals forces between the carbon of the hydrophobic solute and the oxygen in water result in greater ordering in water near the solute.
However, while these views may explain certain thermodynamic or structural observations, they have not been justified experimentally. In the current paper Joze Grdadolnik, Franci Merzel, and Franc Avbelj use vibrational spectroscopy of the oxygen-hydrogen stretching mode to look at relative hydrogen bond strength. By comparing bulk water to water near hydrophobic solute molecules, they were able to confirm that the classical view of the formation of "icebergs" was the best explanation for the hydrophobic effect.
Grdadolnik et al. looked at isotopically decoupled IR oxygen-deuterium (O-D) stretching mode of water near four hydrophobic solvents, methane, ethane, krypton, and xenon. In vibrational spectroscopy a downshift (redshift) indicates that the hydrogen bond strength is enhanced, while the spectral line width indicates whether something is more or less ordered.
Notably, all four of the solutes tested demonstrated a redshift of about 60 cm-1, which is what would be expected for HDO ice and HDO clathrates. This indicates that the hydrogen bonds near the hydrophobic solutes become enhanced relative to bulk water.
The O-D line widths were narrower than bulk water but wider than clathrates. This indicated some structural ordering that is among those water molecules that are near the hydrophobic solute molecules.
Ab initio molecular dynamics simulations were used to investigate the possible structural and electrostatic changes of D2O near methane at various temperatures. These studies showed that the electrostatic potentials of water nearest to the methane molecules were lower than bulk water. Specifically, they noted that four of the nearest water molecules surrounding the tagged water in the first hydration shell interacted more strongly with the tagged water molecule. This particular result seemed to indicate a tendency of hydration water to form locally more stable tetrahedral structures relative to bulk.
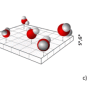
Because hydrogen bonding in water is an electrostatic process, Grdadolnik et al. investigated the relationship between the electric field surrounding a hydrogen bond and the hydrogen bond strength. They identified three distinct classes of water molecules. The first two classes have to do with water molecules located in a fused tetrahedron (shape found in ice).
The third class consisted of intercalating water molecules. They occupy a space that is normally empty in ice. When water molecules are near the solute, methane will occupy these spaces, which causes a bonding structure similar to clathrates. Furthermore, these methane molecules do not shield the electrostatic field the way water molecules do. This results in stronger bonds near the hydrophobic solute.
"The new physical origin of hydrophobicity based on electrostatic screening indicates that hydrophobic and electrostatic interactions in biomolecules are coupled," says Dr. Avbelj. "Such coupling may be very important in biochemical processes. Quantifying this effect might help to improve existing force fields used in atomistic simulations of biomolecules."
More information: Joze Grdadolnik et al. Origin of hydrophobicity and enhanced water hydrogen bond strength near purely hydrophobic solutes, Proceedings of the National Academy of Sciences (2017). DOI: 10.1073/pnas.1612480114
Abstract
Hydrophobicity plays an important role in numerous physicochemical processes from the process of dissolution in water to protein folding, but its origin at the fundamental level is still unclear. The classical view of hydrophobic hydration is that, in the presence of a hydrophobic solute, water forms transient microscopic "icebergs" arising from strengthened water hydrogen bonding, but there is no experimental evidence for enhanced hydrogen bonding and/or icebergs in such solutions. Here, we have used the redshifts and line shapes of the isotopically decoupled IR oxygen–deuterium (O-D) stretching mode of HDO water near small purely hydrophobic solutes (methane, ethane, krypton, and xenon) to study hydrophobicity at the most fundamental level. We present unequivocal and model-free experimental proof for the presence of strengthened water hydrogen bonds near four hydrophobic solutes, matching those in ice and clathrates. The water molecules involved in the enhanced hydrogen bonds display extensive structural ordering resembling that in clathrates. The number of ice-like hydrogen bonds is 10–15 per methane molecule. Ab initio molecular dynamics simulations have confirmed that water molecules in the vicinity of methane form stronger, more numerous, and more tetrahedrally oriented hydrogen bonds than those in bulk water and that their mobility is restricted. We show the absence of intercalating water molecules that cause the electrostatic screening (shielding) of hydrogen bonds in bulk water as the critical element for the enhanced hydrogen bonding around a hydrophobic solute. Our results confirm the classical view of hydrophobic hydration.
Journal information: Proceedings of the National Academy of Sciences
© 2017 Phys.org