Genomic tools for species discovery inflate estimates of species numbers, biologists contend
Increasingly popular techniques that infer species boundaries in animals and plants solely by analyzing genetic differences are flawed and can lead to inflated diversity estimates, according to a new study from two University of Michigan evolutionary biologists.
Lacey Knowles and Jeet Sukumaran investigated the accuracy of inferences made by a mathematical model widely used to quickly determine the boundaries between species without the time-consuming, painstaking process of comparing specimens in museum collections.
They found that the genetic approach, formally known as the multispecies coalescent model, can lead to species estimates that are five to 13 times higher than the true numbers.
Because the species is the fundamental unit for all evolutionary and ecological studies, their findings are expected to have wide-ranging implications, from biodiversity studies to conservation planning. Their results are scheduled for online publication Jan. 30 in Proceedings of the National Academy of Sciences.
"This is an area that has really taken off over the last decade. On its surface, the genomic approach looks like a panacea because it's very fast and doesn't require any kind of taxonomic expertise," said Knowles, a professor in the U-M Department of Ecology and Evolutionary Biology and curator of insects at the university's Museum of Zoology.
"So it's been promoted as a way to speed up inventories of biodiversity by combining the automation of genomics with the statistical power of these models. The only problem is, this method is not doing what we think it is doing, resulting in an overestimate of species numbers."
The U-M researchers say their paper serves as both a warning and a call to action—a warning against reliance on genomic data alone and a call for new methods to improve genomic-based species delimitation approaches.
For now, results from such studies should be considered "at best as tentative hypotheses of species" to be confirmed or rejected through additional analysis using traditional taxonomic methods, such as the physical comparison of museum specimens, according to the authors.
The multispecies coalescent model is widely used to assess understudied populations in known biological hotspots. For example, the approach has been applied to studies of lizards and snakes in southwestern Australian deserts, Amazonian frogs, savanna plants in Brazil and beetles from the Andes.
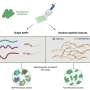
Tissue samples from target organisms are collected in the field—toe pads from desert lizards in various locations, for example—and DNA from the samples is later sequenced in the lab, revealing genetic differences among individuals. The multispecies coalescent model then looks at the genetic differences and attempts to draw boundaries between species.
"Suddenly it seemed like there was a magic bullet. You just have to push a button and you get your species," said Sukumaran, an assistant research scientist in the U-M Department of Ecology and Evolutionary Biology. "But a lot of people got carried away."
Mathematical models are simplified representations of reality and always include assumptions about how the world works. One of the assumptions used to simplify the multispecies coalescent model is that new species form instantaneously after a population of plants or animals becomes geographically isolated.
In reality, not all isolated populations become new species, and the speciation process involves a gradual accumulation of genetic differences over decades, millennia or even millions of years.
"Everyone knows that speciation is not an instantaneous process. But what no one has questioned, until now, is how ignoring that fact changes the story this model is telling us," Sukumaran said. "This paper places that issue front and center."
Sukumaran and Knowles wanted to know what would happen if the multispecies coalescent model was applied to situations in which speciation is a protracted process rather than an instantaneous event.
They used simulated genetic data to compare how the model handled those two scenarios and found that the model overestimates species numbers when it fails to account for the protracted nature of speciation, averaging five to 13 times more estimated species than were actually present in the data.
The inflated species estimates happen because the model misidentifies normal within-species patterns of genetic variation, which biologists call genetic structure, as species boundaries, according to Sukumaran and Knowles. In recent decades, a flood of genomic data collected around the globe from all types of organisms has revealed increasingly finer details in genetic structure, as though biologists had suddenly gained access to a new, more powerful type of microscope.
Paradoxically, this more detailed, higher-resolution view of genomes has made it harder, rather than easier, to distinguish the boundaries between species, according to Knowles and Sukumaran. That's largely because the multispecies coalescent model cannot distinguish the genetic differences found among isolated populations of animals and plants from the true species boundaries, they conclude.
"The irony is that the more genomic data we collect, the less certain we are as to where the species boundaries lie," Knowles said. "Going forward, we are going to need to both improve our models and fall back on alternate—and maybe even more traditional—forms of data to be able to identify species in the age of big data."
More information: Multispecies coalescent delimits structure, not species, PNAS, www.pnas.org/cgi/doi/10.1073/pnas.1607921114
Journal information: Proceedings of the National Academy of Sciences
Provided by University of Michigan