A step toward a UTI treatment that could thwart bacterial drug resistance

This spring, a Pennsylvania woman who developed a urinary tract infection (UTI) became the first patient in the U.S. reported to have a bacterial infection resistant to colistin, an antibiotic of last resort. The case set off alarm bells in the medical field. Now scientists report in ACS' Journal of Medicinal Chemistry a new class of small molecules for fighting the common infection. The way the molecules work could potentially prevent bacteria from developing resistance.
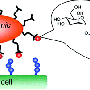
Millions of women in the U.S. every year suffer from UTIs. Antibiotics can treat this painful condition, but with the rise of drug-resistant bacteria, scientists are searching for alternative approaches. James W. Janetka and colleagues' strategy is to develop therapeutic molecules that prevent the bacteria that cause UTI from latching on to key cells on the lining of the urinary tract. Unlike antibiotics, which aim to kill the bacteria, these molecules interfere with the function of a critical factor, FimH, that UTI-causing bacteria make and put on their surface, enabling them to stick to the bladder lining. Such an approach would likely make it more difficult for bacteria to find a workaround. Several teams have designed molecules called O-mannosides that can bind the bacteria, but in animal testing, the compounds appeared to break down too quickly. Janetka's group wanted to address this limitation.
The researchers replaced molecular bonds in O-mannosides with carbon-based linkers to create more stable versions called C-mannosides. Testing in animal models of UTI showed that C-mannosides were more effective at fighting infections than O-mannosides. Ongoing preclinical studies will identify which C-mannoside will move into clinical trials. The resulting candidate could become the first therapeutic treatment against this type of infection, with the potential to thwart bacteria's ability to develop resistance, the researchers say.
More information: Laurel Mydock-McGrane et al. Antivirulence-Mannosides as Antibiotic-Sparing, Oral Therapeutics for Urinary Tract Infections, Journal of Medicinal Chemistry (2016). DOI: 10.1021/acs.jmedchem.6b00948
Abstract
Gram-negative uropathogenic Escherichia coli (UPEC) bacteria are a causative pathogen of urinary tract infections (UTIs). Previously developed antivirulence inhibitors of the type 1 pilus adhesin, FimH, demonstrated oral activity in animal models of UTI but were found to have limited compound exposure due to the metabolic instability of the O-glycosidic bond (O-mannosides). Herein, we disclose that compounds having the O-glycosidic bond replaced with carbon linkages had improved stability and inhibitory activity against FimH. We report on the design, synthesis, and in vivo evaluation of this promising new class of carbon-linked C-mannosides that show improved pharmacokinetic (PK) properties relative to O-mannosides. Interestingly, we found that FimH binding is stereospecifically modulated by hydroxyl substitution on the methylene linker, where the R-hydroxy isomer has a 60-fold increase in potency. This new class of C-mannoside antagonists have significantly increased compound exposure and, as a result, enhanced efficacy in mouse models of acute and chronic UTI.
Journal information: Journal of Medicinal Chemistry
Provided by American Chemical Society