Is evolution predictable?

We are surrounded by microorganisms that adapt in their struggle to survive. In plants and animals, such adaptations can take thousands of years; in microorganisms, they sometimes occur within weeks. To understand such rapid evolution, we need new theoretical frameworks and direct observations of evolutionary dynamics. The Research Group is developing such methods and is applying them to analyze sequence data from influenza and human immunodeficiency virus populations. The results provide insights into the properties of the evolutionary process and make it possible to predict the composition of future virus populations.
Viral evolution
Evolution is generally thought of as a a slow process that is hardly noticable over a period of several years. By contrast, microorganisms often mutate very rapidly and can develop new characteristics in the space of weeks. These microorganisms include many pathogens, and their continuous adaptations can have a dramatic impact on the health of plants, animals and humans, especially when pathogens become resistant to drugs.
The rapid advance of sequencing technology has made it possible to closely observe the evolution and spread of pathogens, for example influenza viruses. Every month, the Global Influenza Surveillance and Response System sequences hundreds of influenza viruses. To model and analyze these data intuitively and in real time, the Research Group led by Richard Neher, together with scientists from Seattle, have set up the nextflu.org website. Fig. 1 shows how the website presents the temporal and geographical spread of influenza virus variants. This type of data analysis can assist regulatory authorities to update seasonal influenza vaccines to match the changing virus populations. Similarly, phylogenies can help to track transmission chains in the case of other viral diseases, for example Ebola, and inform containment efforts.
In addition to monitoring the evolution of pathogens, we can also use rapidly mutating microorganisms to study evolutionary processes that would take millions of years in eukaryotes. The abundance of sequence data from microbial populations provides insights into the driving forces and laws of evolution,. Understanding of this dynamics, in turn, will help us define conditions to slow the adaptation of pathogens. The ability to predict the composition and characteristics of pathogen populations is also within reach. These developments are taking place at the interface between new sequencing technologies, microbiology, bioinformatics and mathematical theory and the modelling of evolutionary processes.

The theory of rapidly mutating populations
Population genetics describes the dynamics of mutations under the influence of natural selection, recombination and a variety of random processes in the life cycle of individuals. It is typically assumed that the majority of all observed mutations have no effect on the phenotype, and that only those mutations occasionally spread that favourably alter the organism's characteristics. However, this assumption does not hold true for microorganisms – and especially not for RNA viruses, the group that includes influenza viruses. Hence, a theory is required that specifically takes into account the dynamics of rapidly adapting populations.
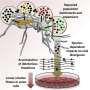
In recent years, the Max Planck Research Group and other scientists have developed models that take into account the dynamics of microbial populations and permit predictions of genetic diversity. Fig. 2 schematically shows the main features of such a model. Individuals in the population have many mutations that differentiate them, and this genetic diversity leads to a diversity of phenotypes, which in turn leads to diversity in replication success, i.e. fitness.
Because fit individuals have more descendants on average, the progenitors of the population typically come from the upper end of the fitness distribution, while the lines of average individuals die out in the long term. Competition among these variants can be mathematically described. This theory provides explicit predictions of the characteristics of the genealogical tree, which can then be compared with sequence data.
The evolution of influenza viruses is predictable
Studying population models has taught us how we can use genome sequences to tell successful, that is fit, viruses from less successful viruses. These predictions are based on patterns in the genealogical tree of the viruses, which can be reconstructed from the sequence data. As successful viruses will dominate in future populations, this insight enables predictions about the composition of future populations.

Reliable predictions are particularly relevant in the case of influenza viruses, as the influenza vaccine must be updated almost every year. Within just a few years, the viruses can change so drastically that outdated vaccines can no longer confer protection. It takes more than six months to produce a new vaccine, so that the decision regarding the composition of a new vaccine has to be made well in advance. Predictions based on a new evolutionary theory relating to influenza viruses can help develop new vaccines quickly.
The Research Group has compared the predictions of their theory with the evolution of influenza viruses between 1995 and 2014. They found that for nearly all the years studied, the method for predicting virus variants agreed closely with the future population that actually occurred. In fact, in many years, the best virus variant was correctly identified. The scientists are currently refining their method in order to provide the World Health Organisation (WHO) with optimum predictions for vaccine compositions.
The evolution of HIV in patients
Another example of rapid evolution is the adaptation of HIV to the host's immune system. This process occurs whenever an individual becomes infected with HIV. To study this process in detail, the Max Planck researchers, together with Jan Albert of the Karolinska Institute in Stockholm have examined archived samples from eleven patients. Five to twelve samples were available from each patient, representing HIV infection from its onset over many years. Each sample contained thousands of HIV genomes, which the scientists decoded using modern sequencing methods. Such time series of samples can be viewed like films, allowing the researchers to track how the virus population changed over time.
Fig. 3 shows the dynamics of mutations in a small segment of the HIV genome within an HIV-positive individual. In this example, ten prevailing mutations are observed within 35 bases. Many other mutations occur as rare variants in fewer than ten percent of the population. Armed with such data, the researchers were able to show that HIV has a kind of optimum sequence, and that around one-third of all mutations represent reversion to the optimum sequence.
These suboptimal variants probably evolved when the virus had to escape the immune system. Such mutations are then reversed in a new host with a different immune system. These and other findings show that HIV populations can readily discover every possible mutation and, from the abundance of possible mutations, almost deterministically select those that best accelerate viral replication. Testing this assumption and developing other methods for predicting virus evolution will continue to occupy the researchers for some time to come.
More information: Richard A. Neher et al. nextflu: real-time tracking of seasonal influenza virus evolution in humans, Bioinformatics (2015). DOI: 10.1093/bioinformatics/btv381
R. A. Neher et al. Genealogies of rapidly adapting populations, Proceedings of the National Academy of Sciences (2012). DOI: 10.1073/pnas.1213113110
Richard A Neher et al. Predicting evolution from the shape of genealogical trees, eLife (2014). DOI: 10.7554/eLife.03568
Fabio Zanini et al. Population genomics of intrapatient HIV-1 evolution, eLife (2015). DOI: 10.7554/eLife.11282
Journal information: Bioinformatics , eLife , Proceedings of the National Academy of Sciences
Provided by Max Planck Society