Simulating in tiny steps gave birth to long-sought-after method
Using computer simulations to predict which drug candidates offer the greatest potential has thus far not been very reliable, because both small drug-like molecules and the amino acids of proteins vary so much in their chemistry. Uppsala researchers have now cunningly managed to develop a method that has proven to be precise, reliable and general.
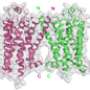
The largest class of human target proteins for drugs are the so-called G-protein-coupled receptors. They are targets for about 40 per cent of all drugs on the market. These receptors are found in the cell membrane and handle the communication between the outside and the inside. When they react to external stimuli, by binding molecules, for example, a structural transformation takes place on the inside that triggers a signalling cascade (see 2012 Nobel Prize in Chemistry).
- In this way these receptors regulate our senses of smell, taste and vision as well as a number of other conditions and feelings, explains Professor Johan Åqvist, who directed the study, which is now being published in the prestigious journal PLOS Computational Biology.
Of the roughly 900 G-linked-protein receptors in humans, today we know the three-dimensional molecular structure of only about twenty. It is important to know this molecular structure when drugs are developed.
The method used today to understand how the receptors function is complicated and time-consuming. First the binding strength of series of molecules is measured (the binding of so-called agonists and antagonists). Then mutations are induced in the receptors in order to see how the binding properties are affected.
- This is both time-consuming and often difficult, because the genetically modified receptors have to be expressed in living cells. With our computational method, the mutation can be created in the computer, and the effect on receptor binding can be calculated with great precision, says Johan Åqvist.
The problem with this type of computer simulation has previously been that the amino acids of the proteins are so different, in terms of size, electrical charge, etc., which has presented problems in the calculations. But when the researchers divided the procedure into a long series of smaller computations, something happened - suddenly they were getting exact and stable results.
The method has now been tested on a neuropeptide receptor and has been shown to be able to predict with great reliability both the effects of mutations and the receptor's ability to bind a series of different molecules. The method also makes it possible to determine whether a three-dimensional structural model of the molecules that are bound to each other is correct.
- The results are brilliant. We believe this has the potential to be extremely useful in drug research. It quite simply makes it easier and faster to find candidates for new drugs. The computational method is also so general that it can be used to study all sorts of other proteins bound to various types of functional molecules, says Johan Åqvist.
More information: Lars Boukharta, Hugo Gutiérrez-de-Terán, Johan Åqvist: Computational Prediction of Alanine Scanning and Ligand Binding Energetics in G-Protein Coupled Receptors. PLOS Computational Biology, Published: April 17, 2014. DOI: 10.1371/journal.pcbi.1003585
Journal information: PLoS Computational Biology
Provided by Uppsala University