A plague in your family: The independent evolution of harmful organisms from one bacterial family

For the first time, researchers have studied the Black Death bacterium's entire family tree to fully understand how some of the family members evolve to become harmful.
Contrary to popular belief, the team found pathogenic members of this bacterial family do not share a recent common disease-causing ancestor, but instead, have followed parallel evolutionary paths to become harmful.
The Yersinia family of bacteria has many sub species, some of which are harmful and others not. Two of the most feared members of this bacterial family are Yersinia pestis, the bacterium responsible for the bubonic plague or the Black Death, and Yersinia enterocolitica, a major cause of gastroenteritis. Previous studies of this family of bacteria have focused on the harmful or pathogenic species, fragmenting our full understanding of the evolution of these species.
"In order to understand how an organism becomes dangerous or pathogenic, we need to understand their non-pathogenic family members to see what makes them different to the pathogenic forms," says Dr Sandra Reuter, first author from the Wellcome Trust Sanger Institute. "Our dataset has allowed us to redefine the family structure of this unique set of bacteria and give us a full view of how an individual bacterial species can become harmful."
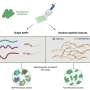
The team sequenced 224 strains of different Yersinia family members from across the world to fully understand of how specific species evolve to become harmful, while others remain harmless.
This unique and detailed dataset describes the parallel evolution of Yersinia pestis and Yersinia enterocolitica; two major disease-causing pathogens in people. The researchers showed that both species, independently of each other, acquired a segment of DNA known as plasmids, as well as the gene ail that allowed them to become pathogenic.
It appears that only these two virulence factors are present in all of the pathogenic Yersinia species.
"Before this study, there was uncertainty about what path these species took to become pathogenic: had they split from a shared common pathogenic ancestor? Or had they evolved independently" says Professor Nicholas Thomson, senior author from the Wellcome Trust Sanger Institute. "What we found were signatures in their genomes which plot the evolutionary path they took.
"Surprisingly they emerged as human pathogens independently from a background of non-pathogenic close relatives. These genetic signatures mark foothold moments of the emergence of these infamous disease-causing bacteria."
The team found that it was not only the acquisition of genes that has proven important to this family of bacteria, but also the loss of genes and the streamlining of metabolic pathways seems to be an important trait for the pathogenic species.
By examining the whole genomes of both the pathogenic and non-pathogenic species, they were able to determine that many of the metabolic functions, lost by the pathogenic species, were ancestral. These functions were probably important for growth in a range of niches, and have been lost rather than gained in specific family lines in the Yersinia family.
"We commonly think bacteria must gain genes to allow them to become pathogens. However, we now know that the loss of genes and the streamlining of the pathogen's metabolic capabilities are key features in the evolution of these disease-causing bacteria," says Dr Alan McNally, senior author from Nottingham Trent University. "This study is shifting our view of the evolution and relationship between species within one family of bacteria."
More information: Reuter S et al. (2014) Parallel independent evolution of pathogenicity within the genus Yersinia. Proceedings of the National Academy of Sciences, 2014. www.pnas.org/cgi/doi/10.1073/pnas.1317161111
Journal information: Proceedings of the National Academy of Sciences
Provided by Wellcome Trust Sanger Institute