The biology of plague: Systems approach used to investigate strains of Yersinia

(Phys.org)—When is the plague not the plague? When it's a different strain of the same bacteria. In two strains of the bacteria genus Yersinia —a highly lethal pathogen and its less-virulent form—scientists performed multi-omic analyses to gain insights how they differ. They found that how the genes in both are expressed contribute to the striking difference in the diseases caused by these pathogens. The research team included scientists from Pacific Northwest National Laboratory, the J. Craig Venter Institute, and the University of Texas Medical Branch.
The work, featured on the cover of Molecular BioSystems, is the next step in ongoing research on Yersinia pestis (YP), which causes plague with a high mortality rate, and Yersinia pseudotuberculosis (YPT), an intestinal pathogen with a modest mortality rate. The bacteria are highly similar with some species-specific differences; however, the molecular causes of their distinct outcomes are poorly understood. The team's analysis revealed that gene and protein expression levels of shared or common virulence-related proteins were both higher in YP than in YPT.
This suggests that adaptation in the regulatory architecture of YP, in addition to the presence of unique genetic material, may contribute to its increased pathogenicity," said the study's lead author Dr. Charles Ansong, PNNL.
The availability of genome sequences for several Yersinia strains, including YPT and both epidemic and non-epidemic YP variants, has provided an opportunity to explore mechanisms responsible for the differences in pathogenicity. Scientists already knew that all human pathogenic Yersinia strains, including YP and YPT, share almost identical genes in plasmids that are essential for virulence.
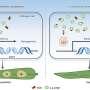
This work highlights the utility of a systems approach incorporating multiple omics measurements and computational analyses to provide novel insights into Yersinia biology; and provides an important resource for the Yersinia research community that should aid the understanding of the markedly different pathogenicities of YP and YPT.
The scientists grew YPT and two forms of YP at temperatures representative of host environments, then sampled them over an 8-hour period. The samples were analyzed using a multi-genome microarray and mass spectrometric methods.
"This experimental design, using multiple omic technologies, was exciting because it allowed us to get a greater grasp of the mechanisms involved in the differences between what have been sometimes called a 'nearly clonal' group of organisms." said PNNL biochemist Dr. Joshua Adkins.
Novel virulence factors predicted from computational analysis of the multi-omics data will be validated in animal models of bubonic plague in follow-on studies. These represent apparent novel therapeutic targets that, with the recent reports of drug-resistant Yersinia strains, are sorely needed.
More information: Ansong, C. et al. A Multi-omic Systems Approach to Elucidating Yersinia Virulence Mechanisms, Molecular BioSystems. DOI: 10.1039/C2MB25287B
Provided by Pacific Northwest National Laboratory