New technique to analyze conformations of complex molecular machines

(Phys.org) —Open, feed, cut. Such is the humdrum life of a motor molecule, the subject of new research at Rice University, that eats and excretes damaged proteins and turns them into harmless peptides for disposal.
The why is obvious: Without these trash bins, the Escherichia coli bacteria they serve would die. And thanks to Rice, the how is becoming clearer.
Biophysicists at Rice used the miniscule machine – a protease called an FtsH-AAA hexameric peptidase – as a model to test calculations that combine genetic and structural data. Their goal is to solve one of the most compelling mysteries in biology: how proteins perform the regulatory mechanisms in cells upon which life depends.
The Rice team of biological physicist José Onuchic and postdoctoral researchers Biman Jana and Faruck Morcos published a new paper on the work this month for a special issue of the Royal Society of Chemistry journal, Physical Chemistry Chemical Physics.
The special issue edited by Rice biophysicist Peter Wolynes and Ruth Nussinov, a researcher at the National Cancer Institute in Frederick, Md., and a professor at the Sackler School of Medicine at Tel Aviv University, pulls together current thinking on how an explosion of data combined with ever more powerful computers is bringing about a second revolution in molecular biology.
The paper describes the Onuchic group's first successful attempt to feed data through their computational technique to describe the complex activity of a large molecular machine formed by proteins. Ultimately, understanding these machines will help researchers design drugs to treat diseases like cancer, the focus of Rice's Center for Theoretical Biological Physics.
"Structural techniques like X-ray crystallography and nuclear magnetic resonance have worked quite well to help us understand how smaller proteins function," Onuchic said. X-rays only take snapshots of constantly moving proteins, he said, "but functional proteins, big protein complexes and molecular machines have multiple conformations.
"Computational models are also useful, but to understand the full dynamics of these large proteins, where a lot of the interesting biology takes place, we have to supplement them with more information," he said.
That information comes from direct coupling analysis (DCA), a statistical tool developed by Morcos and Onuchic with colleagues at the University of California, San Diego, and the Pierre and Marie Curie University. DCA looks at the genetic roots of proteins to see how amino acids – the "beads" in the unfolded protein strands—co-evolved to influence the way a protein folds. Each bead carries an intrinsic energy that contributes to the strand's distinct energy landscape, which dictates how it folds into its functional state.
Even after they fold, proteins are in perpetual motion, acting as catalysts for countless bodily functions. They can combine into larger molecular machines that grab other molecules, "walk" cargoes within a cell or cause muscles to contract.
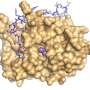
One such biomachine is FtsH, a membrane-bound molecule in E. coli made of six protein copies that form two connected hexagonal rings. The molecule attracts and degrades misfolded proteins and other cellular detritus, pulling them in through one ring, which closes like the shutter of a camera and traps the proteins. They are cut apart as they exit through the other ring.
Through molecular simulations using structure-based models and the discovery via DCA of likely couplings in the genetic source of the proteins, the Rice team found evidence to support the hypothesis of a "paddling" mechanism in the molecule that Morcos described as a collapse of the two rings once trash found its way inside.
"First the ring pore closes to grab the protein; then the molecule flattens," he said. "Then when the motor is flat, the rings open to release the peptides and the molecule expands again to restart the cycle."
Key to the success of DCA is the realization that amino acid mutations represent contacts that co-evolve for specific purposes. The contact maps created by DCA can reveal previously unknown details to model transitions between functional states, like the paddling in FtsH, Onuchic said.
"We can look at the evolutionary tree of these proteins and see which pairs of amino acids changed together. We then assume these are contacts," he said. "Through DCA, Faruck uses a lot of physics to understand when two amino acids can act directly or indirectly, and separate the two. Then we predict how coupled they are, and the higher the probability, the more evidence that these are real contacts."
DCA would do little without the flood of data available since the ability to scan entire genomes became possible, and even commonplace, in recent decades. Advances in the century-old art of crystallography are making better structure-based models available as well.
"Even if the mathematical framework was ready and we had crystallographic data for this motor protein in the 1990s, there weren't enough sequences available until the 2000s," Morcos said. "Now we have all the pieces converging."
He said understanding essential motor proteins in bacteria will be important as researchers begin to apply DCA to advance human health. "For us, the most exciting part is that we're now able to tackle really big systems," Morcos said.
More information: "From structure to function: the convergence of structure based models and co-evolutionary information." Biman Jana,ab, Faruck Morcosa, José N. Onuchic. Phys. Chem. Chem. Phys., 2014,16, 6496-6507. DOI: 10.1039/C3CP55275F. Received 14 Dec 2013, Accepted 10 Feb 2014. First published online 07 Mar 2014
Journal information: Physical Chemistry Chemical Physics
Provided by Rice University