Shifting evolution into reverse promises cheaper, greener way to make new drugs
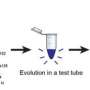
A proof-of-concept experiment has shown that, by shifting evolution into reverse, it may be possible to use 'green chemistry' to make a number of costly synthetic drugs as easily and cheaply as brewing beer.
This alternative approach to creating artificial organic molecules, called bioretrosynthesis, was first proposed four years ago by Brian Bachmann, associate professor of chemistry at Vanderbilt University. Now Bachmann and a team of collaborators report that they have succeeded in using the method to produce the HIV drug didanosine.
The proof of concept experiment is described in a paper published online March 23 by the journal Nature Chemical Biology.
"These days synthetic chemists can make almost any molecule imaginable in an academic laboratory setting," said Bachmann. "But they can't always make them cheaply or in large quantities. Using bioretrosynthesis, it is theoretically possible to make almost any organic molecule out of simple sugars."
Putting natural selection to use in this novel fashion has another potential advantage. "We really need a green alternative to the traditional approach to making chemicals. Bioretrosynthesis offers a method to develop environmentally friendly manufacturing processes because it relies on enzymes – the biological catalysts that make life possible – instead of the high temperatures and pressures, toxic metals, strong acids and bases frequently required by synthetic chemistry," he said.
Normally, both evolution and synthetic chemistry proceed from the simple to the complex. Small molecules are combined and modified to make larger and more complex molecules that perform specific functions. Bioretrosynthesis works in the opposite direction. It starts with the final, desired product and then uses natural selection to produce a series of specialized enzymes that can make the final product out of a chain of chemical reactions that begin with simple, commonly available compounds.
Bachmann got the idea of applying natural selection in reverse from the retro-evolution hypothesis proposed in 1945 by the late Caltech geneticist Norman Horowitz. Horowitz envisioned an early stage in the development of life where early organisms were swimming in a primordial soup rich in organic material. In this environment, imagine that one of the species finds a use for the complex chemical compound A that gives it a competitive advantage. As a result, its population expands, consuming more and more compound A. Everything goes well until compound A becomes scarce. When that happens, individuals who develop an enzyme that allows them to substitute the still plentiful compound B for the scarce compound A gain a reproductive advantage and continue to grow while those who remain dependent on compound A die out. And so it goes until many generations later the survivors have developed multi-step chemical pathways to produce the molecules that they need to survive from the molecules available in their environment.
To test Bachmann's retro approach, the Vanderbilt chemists first identified the drug that they wanted to produce – in this case didanosine, an anti-HIV drug sold under the trade names of Videx and Videx EC that is very costly to manufacture. Then they identified a similar "precursor" molecule that can be converted into didanosine when it is subject to a specific chemical transformation along with an enzyme capable of producing the type of transformation required.
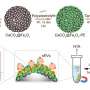
Once they identified the enzyme, the researchers made use of the power of natural selection by making thousands of copies of the gene that makes the enzyme using a special copying technique that introduces random mutations.
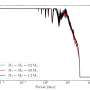
The mutant genes were transferred into the gut bacteria E. coli in order to produce the mutant enzymes and placed into different "wells." After the cells were broken open and the contents mixed with the precursor compound, the amount of didanosine, in each well was measured. The researchers selected the enzyme that produced the greatest amount of the desired drug and then made enough of this optimized enzyme for the next step.
Next the researchers identified a second precursor – an even simpler molecule that could be chemical converted into the first precursor – and an associated transformative enzyme. Again they made thousands of mutated versions of the transformative enzyme's gene, inserted them in E. coli, put them in wells, broke open the cells and mixed the content with the optimized enzyme and second precursor. Once again, they tested all the wells for the anti-HIV drug. The well with the highest level of didanosine was the one in which the mutant enzyme was most effective in making the first precursor, which the optimized enzyme then converted into didanosine. This gave them a second optimized enzyme. The researchers carried out this reverse selection process three times, until they could make didanosine out a simple and inexpensive sugar named dideoxyribose.
One of the key technical challenges was rapidly determining the three-dimensional structures of the enzymes that were generated during the evolutionary process. Associate Professor of Pharmacology Tina Iverson provided this capability. Her team analyzed the laboratory-evolved enzymes after each round of mutagenesis and identified how the structural changes caused by the mutations improved the enzyme's ability to produce the desired transformation.
This information helped the collaborators figure out why some mutant enzymes did a better job at producing the desired compounds than others, which guided their choices about the areas of the precursor proteins to target.
The proof-of-concept experiment was performed in vitro instead of in living cells to keep things simple. However, the ultimate goal is to use the approach to produce artificial compounds by fermentation.
More information: Bioretrosynthetic construction of a didanosine biosynthetic pathway, Nature Chemical Biology, DOI: 10.1038/nchembio.1494
Journal information: Nature Chemical Biology
Provided by Vanderbilt University