Researchers take next step toward enhancing artificial proteins

(Phys.org)—A team of researchers has developed a method to successfully predict the structures of artificial proteins, a breakthrough that could yield valuable methods for making pharmaceuticals and other chemicals that require precise assembly of complex structures.
The work, which appears in the Proceedings of the National Academy of Sciences (PNAS), was conducted by researchers at NYU, the U.S. Department of Energy's Lawrence Berkeley National Laboratory (Berkeley Lab), Stony Brook University, and Temple University.
The structures of natural proteins define their complex functions. Based on interactions between their amino acids, proteins can fold and twist into distinct, chemically directed shapes. The resulting structure dictates the proteins' actions in the body, so accurate modeling of structure is vital to understanding their functions.
Peptoids, or synthetic proteins, follow similar design rules. Because peptoids are less vulnerable to chemical or metabolic breakdown than are proteins, they hold promise for pharmaceuticals and materials. Moreover, scientists can now build and manipulate peptoid molecules with great precision. But to design peptoids for a specific function, researchers need to first untangle the complex relationship between a peptoid's composition and its folded structure.
Past efforts to predict protein structure have had limited success, but the research team for the PNAS study demonstrated that a computer modeling approach similar to one used to predict protein structures can accurately predict peptoid conformations as well.
"The research was carried out by a remarkable, interdisciplinary team of scientists," said Kent Kirshenbaum, an associate professor in NYU's Department of Chemistry and one of the study's co-authors. "Some of the team have worked together on this truly difficult problem for almost 20 years. The researchers include both experimentalists and theorists who have been able to guide one another in discovering how these peptoid molecules fold."
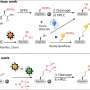
The research team, which also included Richard Bonneau, an associate professor in NYU's Department of Biology, devised an innovative approach to make accurate predictions of peptoid folding—a "blind structure prediction" challenge. This self-assessment technique allows scientists to test the fidelity of their computational models by predicting the three-dimensional structure of a known molecule and then comparing their proposed structure to the X-ray crystallography results.
A combined experimental and computational method was employed by the peptoid team in an effort to advance the computational design of peptoid structures. X-ray crystal structures for three peptoid molecules, two small linear molecules and one large cyclic molecule, were simultaneously determined, but not disclosed to the theoretical modelers. The experimentalists then used a combination of two simulation techniques, Replica Exchange Molecular Dynamics (REMD) simulation and Quantum Mechanical refinement (QM). REMD can efficiently predict the preferred general conformations, and the QM calculations further refine the conformational prediction. In combination, these two calculations accurately define the physical structures of molecules.
The proposed structural predictions of the peptoid molecules did exceedingly well at calculating the actual folded conformations, suggesting that reliable structure prediction for complex three-dimensional folds is within reach and marking an enormous step forward on the path to reliable and efficient computational peptoid design.
Journal information: Proceedings of the National Academy of Sciences
Provided by New York University