Sequencing technique unveiling the realm of viral mutations

Researchers at A*STAR have devised a sequencing technique that can track specific viral variants produced when viruses such as hepatitis B rapidly mutate within individual patients. The breakthrough allows an unprecedented view of evolving virus population structures, and could help in the creation of new drugs that prevent the development of strains resistant to drugs and immune responses.
Viruses mutate through small alterations within each genome to generate single-nucleotide variants, or SNVs. Sets of single-nucleotide variants that occur on a viral genome are known as haplotypes. These mutations vary from one host to the next, creating copies of the viral genome that are all slightly different from one another. Scientists believe that mutated genomes have a role to play in viral replication.
"Deep DNA sequencing has revolutionized the study of viral evolution," states William Burkholder, who led the team from the A*STAR Institute of Molecular and Cell Biology alongside scientists from Singapore, the United Kingdom and the United States. "However, it is not sensitive enough to pick up rare SNVs, find co-occurring mutations on the same genome, or organize the mutations into distinct genetic groups within the viral population. Our technique aims to resolve these issues."
The team refined existing deep sequencing techniques by creating what they call Barcode-directed Assembly for Extra-long Sequences (or BAsE-Seq) using a low-cost, short-read sequencer. The system pinpoints exactly which mutations relate to which viral genome using barcode tagging, and can help identify long viral haplotypes with increased accuracy and sensitivity.
"The trick is to generate short fragments of DNA from long DNA templates while keeping track of where each fragment came from," explains Burkholder. "To do this, we assigned each fragment a barcode with the 'address' of its original molecule. This meant that we could reconstruct the complete DNA sequence once we had found the mutations on each fragment."
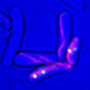
Analysis of a hepatitis B virus clinical sample using BAsE-Seq allowed the team to obtain over 9,000 viral haplotypes, giving an unprecedented view of the overall virus population structure. Based on these data, the researchers created a phylogenetic tree that illustrates the diversity of the virus within the patient sample (see image). They successfully identified unique haplotypes occurring with frequencies of 0.4 per cent and above in each patient.
"BAsE-Seq is a powerful, cost-effective approach for studying the population dynamics of DNA viruses," states Burkholder. "We will extend the technique for other viral applications in future."
More information: "A method for obtaining long viral haplotypes from short sequence reads." Genome Biology 15, 517 (2014). dx.doi.org/10.1186/s13059-014-0517-9
Journal information: Genome Biology