Research finds that malaria parasites are unlikely to jump to humans
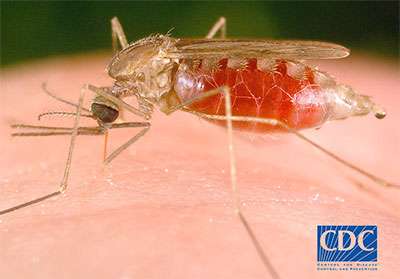
In recent years, public health experts have increasingly explored the idea of eliminating the most dangerous malaria-causing parasite. But they have questioned whether getting rid of this species, called Plasmodium falciparum, would allow other species of the parasite to simply jump into the gap and start infecting humans with malaria.
Now, a new study led by a researcher at the University of Maryland School of Medicine indicates it is very unlikely that Plasmodium species that infect other animals—such as apes, birds and reptiles—would cross over easily to humans. Using sophisticated genetic analysis, Joana C. Silva, PhD, found evidence showing that five other common Plasmodium species have not changed which animals they infect for at least 3 million years.
Malaria is a leading cause of disease and death throughout the world; every year it infects over 200 million people, and causes more than half a million deaths. In parts of sub-Saharan Africa and South Asia, the disease is common and causes enormous suffering and hardship.
The new study was published last month in the journal Molecular Biology and Evolution. Dr. Silva, an assistant professor in the Department of Microbiology and Immunology and at the Institute for Genome Sciences at the UM SOM, was the lead author.
"This is a key question - how likely are these parasite species to jump to humans?" says Dr. Silva. "And according to our results, "host switching" by malaria-causing parasites is not at all a common event, on an evolutionary time scale."
More than 200 Plasmodium species have been identified. Plasmodium falciparum is the most lethal of the five that are known to infect humans. Researchers are examining new approaches to reduce or eliminate Plasmodium falciparum by developing vaccines against it, for example, or spreading a bacterium that kills the mosquitoes that carry it. But some scientists have expressed concern that Plasmodium falciparum's ecological niche might be quickly filled by other Plasmodium species.
Dr. Silva and her co-authors looked at hundreds of genes spread across five different species of Plasmodium. Their goal was to discover how closely related the genes were—in effect, how long ago they had diverged from each other. If they had separated recently, it was more likely that they could jump from infecting one species to another.
To get their results, Dr. Silva and her colleagues developed a new statistical approach to determine when Plasmodium species split off from one another. The new method uses molecular data from thousands of genes; current techniques, by contrast, use at most sequences from dozens. This new approach is not only more reliable, but also faster.
"This is exciting research that has powerful public health implications," said Dean E. Albert Reece, MD, PhD, MBA, who is also the vice president for Medical Affairs, University of Maryland, and the John Z. and Akiko K. Bowers Distinguished Professor and Dean of the School of Medicine. "It is particularly interesting to see the application of 'big data' and genetic analysis increasingly being used to help solve the world's most critical health problems."
The research team included scientists and statisticians from the National Center for Biotechnology Information at the National Institutes of Health and the Applied Mathematics & Statistics, and Scientific Computation program at the University of Maryland, College Park (UMCP).
"This is an exciting integration of mathematics and genetics," said David Harris, a researcher in the UMCP Applied Mathematics & Statistics and Scientific Computation program, who developed the statistical methods used in this research. "It's great to be able to use mathematics in a way that has the potential to inform practical policy decisions."
Journal information: Molecular Biology and Evolution
Provided by University of Maryland