Gene activity and transcript patterns visualized for the first time in thousands of single cells

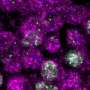
Biologists of the University of Zurich have developed a method to visualize the activity of genes in single cells. The method is so efficient that, for the first time, a thousand genes can be studied in parallel in ten thousand single human cells. Applications lie in fields of basic research and medical diagnostics. The new method shows that the activity of genes, and the spatial organization of the resulting transcript molecules, strongly vary between single cells.
Whenever cells activate a gene, they produce gene specific transcript molecules, which make the function of the gene available to the cell. The measurement of gene activity is a routine activity in medical diagnostics, especially in cancer medicine. Today's technologies determine the activity of genes by measuring the amount of transcript molecules. However, these technologies can neither measure the amount of transcript molecules of one thousand genes in ten thousand single cells, nor the spatial organization of transcript molecules within a single cell. The fully automated procedure, developed by biologists of the University of Zurich under the supervision of Prof. Lucas Pelkmans, allows, for the first time, a parallel measurement of the amount and spatial organization of single transcript molecules in ten thousands single cells. The results, which were recently published in the scientific journal Nature Methods, provide completely novel insights into the variability of gene activity of single cells.
Robots, a fluorescence microscope and a supercomputer
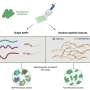
The method developed by Pelkmans' PhD students Nico Battich and Thomas Stoeger is based upon the combination of robots, an automated fluorescence microscope and a supercomputer. "When genes become active, specific transcript molecules are produced. We can stain them with the help of a robot", explains Stoeger. Subsequently, fluorescence microscope images of brightly glowing transcript molecules are generated. Those images were analyzed with the supercomputer Brutus, of the ETH Zurich. With this method, one thousand human genes can be studied in ten thousand single cells. According to Pelkmans, the advantages of this method are the high number of single cells and the possibility to study, for the first time, the spatial organization of the transcript molecules of many genes.
. Credit: Pelkmans Lab")
New insights into the spatial organization of transcript molecules
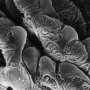
The analysis of the new data shows that individual cells distinguish themselves in the activity of their genes. While the scientists had been suspecting a high variability in the amount of transcript molecules, they were surprised to discover a strong variability in the spatial organization of transcript molecules within single cells and between multiple single cells. The transcript molecules adapted distinctive patterns.
. Credit: Pelkmans Lab")
"We realized that genes with a similar function also have a similar variability in the transcript patterns," explains Battich. "This similarity exceeds the variability in the amount of transcript molecules, and allows us to predict the function of individual genes." The scientists suspect that transcript patterns are a countermeasure against the variability in the amount of transcript molecules. Thus, such patterns would be responsible for the robustness of processes within a cell.
. Credit: Pelkmans Lab")
The importance of these new insights was summarized by Pelkmans: "Our method will be of importance to basic research and the understanding of cancer tumors because it allows us to map the activity of genes within single tumor cells."
More information: Nico Battich, Thomas Stoeger, Lucas Pelkmans. Image-based transcriptomics in thousands of single human cells at single-molecule resolution. Nature Methods. DOI: 10.1038/nmeth.2657
Journal information: Nature Methods
Provided by University of Zurich