May 7, 2013 report
Sequencing reveals complex history of amphibian-killing fungus

(Phys.org) —One of the biggest threats facing amphibian species is the disease chytridiomycosis, which is caused by a fungus known as Batrachochytrium dendrobatidis (Bd). An understanding of the evolutionary history of microbial pathogens, like Bd, is critical to predicting disease outbreaks and causes of shifts in virulence. The sudden appearance of Bd worldwide suggests a recent introduction into the affected areas, likely facilitated by the international movement of amphibians. A new paper published yesterday in PNAS reveals that the evolutionary history of Bd significantly predates recent outbreaks, and is much deeper and more complex than previously thought. Using whole-genome sequencing from a global panel of Bd isolates, researchers suggest that Bd likely originated somewhere between 10,000 and 40,000 years ago, but could be as old as 100,000 years.
The decline in amphibian populations due to Bd is concerning since they play an essential role in food webs, and have broad aesthetic and medicinal value. Bd infects hundreds of species of amphibians and kills by disrupting the integrity of their skin. Earlier, but limited, studies found little genetic differentiation in Bd, with no clear geographic signal. This would be consistent with a recent, rapid spread of a novel disease agent. The new study in PNAS, is an extensive collaboration including researchers from the US, Mexico, Argentina, Columbia and Australia. It makes use of whole-genome sequencing to show that the geographic lineage of Bd is much more widespread and diverse than had been assumed.
If Bd were largely endemic, researchers would expect deep phylogenetic structure in the Bd tree corresponding to locality or host association. If instead, Bd were entirely novel, they would expect a shallow and comb-like tree topology. To infer the relationships in the Bd tree, the researchers used 29 isolates sequenced to depth of 24x, and 101,000 SNPs (single nucleotide polymorphisms). What they found was a history that was both novel and endemic. Generally, isolates that shared a common geographic or host association did not cluster within clades. Similarly, many closely related isolates did not share a common geography.
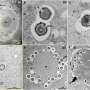
, the chytrid fungus responsible for many amphibian declines. Photograph by Erica Bree Rosenblum, University of California at Berkeley. Below: Hypsiboas polytaenius from Brazil, one of the many amphibian species infected by Bd. Photograph by David Rodriguez, Cornell University. Credit: Erica Bree Rosenblum")
The authors suggest that these findings do not reflect ancient geographic structure, but instead, are likely explained by invasion and subsequent evolution of clones in particular regions. By comparing their genomic set with that of a non-Bd set, the researchers were able to identify lineages that were more basal in the Bd tree and determine association with particular hosts and regions. They believe that the basal lineage is traceable to Brazil, which contained the most ancestral variation. Bd has been reported in a large number of Brazilian frog species although the mass die offs found elsewhere have not been observed there.
The researchers applied a molecular clock to estimate the age of key nodes within the phylogenetic tree. The dates provided are dependent on an assumed rate of evolution under a strict clock (0.0081 substitutions per site per million years) and a tree prior derived from the constant population-size coalescent. It should be noted that these kinds of model assumptions have received some valid criticisms in past and should only serve as estimates. The researchers also note that although sexual reproduction has never been observed in Bd, sexual recombination and hybridization have been proposed as important mechanisms in its evolutionary history.
For the future, the researchers hope to focus on some of the less sampled parts of the world, and include additional host species. They also hope to link phylogenetic variation with functional variation. Evidence from the lab has suggested that some more recent isolates may be more deadly than the early-diverging isolates, particularly those from South Africa. Because virulence is an emergent property of the host-microbe-environment interaction, comparisons among isolates will be difficult. Controlled experiments and consistent assays will be needed to piece together parts of the larger Bd puzzle.
More information: Complex history of the amphibian-killing chytrid fungus revealed with genome resequencing data, PNAS, Published online before print May 6, 2013, doi: 10.1073/pnas.1300130110
Abstract
Understanding the evolutionary history of microbial pathogens is critical for mitigating the impacts of emerging infectious diseases on economically and ecologically important host species. We used a genome resequencing approach to resolve the evolutionary history of an important microbial pathogen, the chytrid Batrachochytrium dendrobatidis (Bd), which has been implicated in amphibian declines worldwide. We sequenced the genomes of 29 isolates of Bd from around the world, with an emphasis on North, Central, and South America because of the devastating effect that Bd has had on amphibian populations in the New World. We found a substantial amount of evolutionary complexity in Bd with deep phylogenetic diversity that predates observed global amphibian declines. By investigating the entire genome, we found that even the most recently evolved Bd clade (termed the global panzootic lineage) contained more genetic variation than previously reported. We also found dramatic differences among isolates and among genomic regions in chromosomal copy number and patterns of heterozygosity, suggesting complex and heterogeneous genome dynamics. Finally, we report evidence for selection acting on the Bd genome, supporting the hypothesis that protease genes are important in evolutionary transitions in this group. Bd is considered an emerging pathogen because of its recent effects on amphibians, but our data indicate that it has a complex evolutionary history that predates recent disease outbreaks. Therefore, it is important to consider the contemporary effects of Bd in a broader evolutionary context and identify specific mechanisms that may have led to shifts in virulence in this system.
Journal information: Proceedings of the National Academy of Sciences
© 2013 Phys.org