In race to predict protein structure, computers take lead
A flood of data is emerging from genome research, including sequence data on proteins. To help science keep pace with this flow of knowledge, computer scientists, biophysicists and biochemists across the world have been developing advanced technologies to help derive accurately and quickly the three-dimensional structure of proteins from this data.
At a competition that has been called the "Olympic games of protein structure prediction," two teams of computer scientists at the University of Missouri were ranked among the best in the world. Their new, faster and more accurate protein structure prediction servers will help scientists better determine the function of proteins in cells.
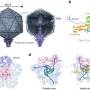
Proteins serve many functions in cells. Some proteins make hair strong and flexible, while others help digest food and contribute to almost every function needed for life. What function a protein serves is determined by its compact three-dimensional shape dictated by a unique sequence of amino acids encoded by the genome. If a protein gets misshapen or misfolded, it stops working properly. In humans, the accumulation of misfolded proteins is linked to a number of disorders, including Parkinson's disease, cancer and diabetes.
"Given the importance of protein structure to all biological processes, the ability to accurately predict protein structure from sequence data is one of the most challenging problems in biology today," said Jianlin Cheng, assistant professor of computer science in the MU College of Engineering.
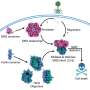
It also is a problem that can be solved with simulations running on computer servers. Now, research groups worldwide are in a race to see who can develop the best server.
Critical Assessment of Techniques for Protein Structure Prediction (CASP) is a competition that pits computer modeling designed by groups from around the world to see whose method comes closest to structures determined in the laboratory. The goal is to provide a rigorous, peer-reviewed test of the accuracy of current computational protein structure prediction methods.
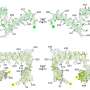
Results from the most recent competition, CASP8, were recently announced. Among the prediction methods ranked best in the world in both template-free and template-based categories were MULTICOM and MUFOLD, both designed by teams of computer scientists at MU. The two prediction categories differ by whether the unsolved protein sequence is generated based on known structures or deduced solely from sequence data.
Both teams predicted the folding of 128 proteins from a number of different species, including those from bacteria, viruses, and both single- and multi-celled organisms.
The MULTICOM team, led by Cheng, included Zheng Wang, a graduate student in computer science; and Allison Tegge, a graduate student in bioinformatics.
The MUFOLD team included Dong Xu, professor of computer science; Yi Shang, professor of computer science; and Ioan Kosztin, an associate professor of physics. Bogdan Barz, a graduate student in physics; Zhiquan He and Qingguo Wang, graduate students in computer science; and Jingfen Zhang, a postdoctoral fellow also were members of the prediction team.
Cheng and Xu, both members of the MU Interdisciplinary Plant Group and investigators in the Christopher S. Bond Life Sciences Center, are using their technologies to help plant scientists determine the structure and function of proteins in a number of important crop plants, including corn and soybean.
Source: University of Missouri-Columbia