This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
proofread
Molecular simulations of ammonia mixtures support search for renewable fuels

Ammonia (NH3) is an important molecule with many applications. The end product of the famed Haber–Bosch process, it is commonly synthesized to capture nitrogen for fertilizers, and is used for refrigeration, in cleaning products, and in the production of pharmaceuticals. Recently, this modest molecule has also attracted interest as a potential resource for addressing one of today's most pressing challenges—the need for reliable and abundant renewable fuels.
Ammonia is stable and safe to handle, is combustible, and contains the largest fraction of hydrogen of any molecule except for pure hydrogen itself. These factors promise to make it a feasible alternative to the carbon-based energy carriers that are driving climate change. Research has begun to explore how ammonia could be used to directly power engines, gas turbines, and hydrogen fuel cells, for example. It is also believed that ammonia could be used to store energy for times when other renewables like wind and solar power cannot meet demand.
Much is known about ammonia, but this interest in using it as a fuel has initiated a search for new ammonia technologies. This has, in turn, led to an increased need among chemical engineers for accurate data describing ammonia's fundamental thermodynamic properties. Such properties include a wide variety of measurable traits such as phase equilibria, density, or heat capacity, for example, that characterize physical systems and determine how chemical processes work. In the case of ammonia, engineers would also like to have better knowledge of how such properties change when mixing ammonia with other molecules. Such knowledge could help them to optimize processes and operating conditions.
Dr. Jadran Vrabec, currently the director of the Institute for Process Sciences at the Technical University of Berlin, has spent much of his career using high-performance computing (HPC) to investigate thermodynamic properties at the molecular level. "Thermodynamic properties are 100% determined by molecular interactions," he explains. "And because these interactions happen so fast and at such a small scale, it is only possible to study them by performing large simulations using supercomputers."
In a recent paper published in the Journal of Chemical & Engineering Data, he and co-author Erich Mace of the TU Berlin report on the results of simulations focused on the thermodynamic properties of mixtures containing ammonia. Produced using the Hawk supercomputer at the High-Performance Computing Center Stuttgart (HLRS), their results add valuable data that could support the development of new applications of ammonia. The results could also help to assess the accuracy of other existing data, ensuring that engineers have the best available information for working with the substance.
. DOI: 10.1021/acs.jced.3c00327")
Large-scale simulations provide unique insights into thermodynamic properties
Vrabec is a longtime user of HLRS supercomputing resources for molecular dynamics and Monte Carlo simulations. His approach relies on concepts of thermodynamics that were first articulated by Ludwig Boltzmann in the 19th century, but only became practical to apply in the 1950s with the arrival of the first computers. Since then, the field has advanced in parallel with the development of larger and faster supercomputers, to the point that Vrabec's simulations now track the individual motions and interactions of billions or even trillions of molecules simultaneously. Using software his lab developed to selectively capture data of interest, he can then study the molecules' thermodynamic properties.
Vrabec uses two simulation codes called ms2 and ls1, which he has developed and optimized over the course of a long and fruitful collaboration with HLRS staff members Martin Bernreuther and Christoph Niethammer. In 2019 the team even set a world record for the largest molecular system ever simulated using molecular dynamics methods. Using ls1, they efficiently scaled their code to a system of 21 trillion atoms in which every individual molecule and its interactions with other molecules could be tracked.
In the recent work on ammonia, Mace and Vrabec performed molecular dynamics and Monte Carlo simulations using ms2 to investigate five commonly used mixtures involving ammonia in chemical engineering processes: argon–ammonia, methane–ammonia, hydrogen–ammonia, nitrogen–ammonia, and oxygen–ammonia. For each mixture the simulations generated data describing the vapor-liquid equilibrium (VLE)—a measurement of the distribution of molecules in a system across the vapor or liquid phases—for a wide range of temperatures and pressures.
In their paper, Mace and Vrabec point out that VLE data is often used in developing equations of state for industrial fluids; that is, the data can be used to predict the state of matter under different physical conditions due to changes in temperature, pressure, volume, or composition. Such information is essential for determining optimal mixtures and working conditions in industrial applications.
Vrabec's molecular simulations are particularly valuable because they can be used to investigate a much wider range of scales than is possible using experimental approaches.
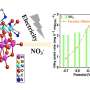
"In our simulations, we provided measurements of thermodynamic properties even up to pressures of 50 megapascals. This is 500 times our ambient air pressure," Vrabec remarks. "Although data for ammonia mixtures have been gathered for more than a century, the data coverage is surprisingly narrow. The reason is that the effort to measure it experimentally is prohibitively huge. It would require expensive special equipment that would be dangerous to operate. In computer simulations, we can get results safely and relatively inexpensively." His methods also provide a comparable level of accuracy to that of experimental approaches in ranges where experimental data is available.
 correspond well to other experimental data, and reveal outliers in experimental data (seen, for example, in the red diamonds for Kaminishi's 1961 results in the lower half of the top left figure) that are likely to be inaccurate. Credit: Journal of Chemical & Engineering Data (2023). DOI: 10.1021/acs.jced.3c00327")
Better data for ammonia research
When Mace and Vrabec analyzed their simulation data, they showed that although ammonia is a component in all five systems they studied, the resulting graphs of VLE values look dramatically different for different molecular mixtures. According to Vrabec, "The phase behavior of different mixtures is strongly determined by the interactions among the molecules in the system. You need to understand these properties if you are interested in working with ammonia mixtures."
The paper and its supplementary data offer more than 400 new data points for each mixture they studied. Using Hawk, they were able to produce the results of each mixture within just a few days of computing time. The results will be of particular value for extreme, difficult-to-study conditions for which little data is available, and could help engineers to identify sweet spots where conditions would be optimal for efficient ammonia processing.
The study included both new simulation data and previously published data from the scientific literature, enabling Mace and Vrabec to compare their results with other existing datasets of VLE values. In most situations, their results corresponded closely with those of previous studies. In some cases, however, they identified significant divergences between their results and other research groups' experimentally generated measurements and predictions. The authors attribute these discrepancies to limitations or inaccuracies in the corresponding experimental methods. They also suggest that specific experimental data sources should be used with caution in future research or chemical engineering applications.
Vrabec says that in recent work, he has focused primarily on simulating thermodynamic properties of molecular systems, generally at the sub-micrometer scale. Despite the many orders of magnitude that lie between this scale and the level of observable processes, accurate methods exist for translating these molecular-level insights into useful real-world predictions.
As supercomputers grow larger, however, he anticipates that it might also become possible to simulate not just properties but also thermodynamic processes using boundary conditions that are close to real-world applications. Increased HPC performance could produce more accurate results about dynamic phenomena with a better signal-to-noise ratio.
In the meantime, though, his team's results demonstrate the value of molecular dynamics and Monte Carlo simulation using high-performance computing, and will provide new understanding of phase behavior that engineers can use to develop new ammonia-based technologies.
More information: Erich J. Mace et al, High-Pressure Fluid-Phase Equilibria and Henry's Constants of Supercritical Gases in Ammonia, Journal of Chemical & Engineering Data (2023). DOI: 10.1021/acs.jced.3c00327
Provided by Gauss Centre for Supercomputing