This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
trusted source
proofread
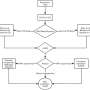
Inferring causative microbial features from metagenomic data of limited samples
. DOI: 10.1016/j.scib.2023.10.024")
Increasing evidence has shown an association between gut microbiota and numerous diseases inferred by metagemomic (MWAS), indicating the microbiota as one of the most promising and effective strategies to control these diseases. However, inferring causalities and strong associations from high-dimensional data are very challenging, leading to low concordance in causal microbe identification between metagenomic studies.
Although great efforts have been made to effectively control the numerous confounding cofactors that make the human microbiota notoriously complex and highly variable, there is still a long way to go when considering the individual heterogeneity in cross-sectional studies.
In a new study led by Prof. Yu Kang of the CAS Key Laboratory of Genome Sciences and Information, Beijing Institute of Genomics Chinese Academy of Sciences & China National Center for Bioinformation, a team of researchers has developed an open-access tool, Virtual Twins (VTwins), which significantly improves the identification of disease-causal microbes from complex metagenomic data.
The research is published in the journal Science Bulletin.
The innovative approach adopted by VTwins is inspired by twin studies in genetic research mimicking the twin samples, which can perfectly control the highly variable genetic background, remarkably reduce the required sample size, and often achieve success in identifying disease-causative genetic variations.
The researchers select paired samples of distinct phenotypes but matched taxonomical profile to reconstruct a new "twin" cohort from the original group cohort. By this simple transition, VTwins is able to effectively control the highly variable metagenomic confounding factors and achieve high significance in the subsequent statistical tests for paired samples.
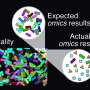
Performance evaluation of VTwins using both simulated and empirical metagenomic datasets demonstrated its superior performance in identifying causative features, even with reduced sample sizes. Furthermore, VTwins was benchmarked against 16 other software and validated for its effectiveness and applicability.
This tool is particularly proficient in controlling non-relevant confounding features and minimizing background noise, which are common challenges in metagenomic research. This reduces 10-fold the sample size required for identifying disease-associated microbial features, making VTwins an invaluable tool in high-dimension data analysis in the big data era.
As metagenomic research continues to draw attention to the relationships between the human microbiota and a range of disease conditions, tools like VTwins are critical for insightful understanding of disease pathogenesis.
VTwins is open access and available for immediate use online.
More information: Qingren Meng et al, VTwins: inferring causative microbial features from metagenomic data of limited samples, Science Bulletin (2023). DOI: 10.1016/j.scib.2023.10.024
Provided by Science China Press