This article has been reviewed according to Science X's editorial process and policies. Editors have highlighted the following attributes while ensuring the content's credibility:
fact-checked
peer-reviewed publication
trusted source
proofread
Mammalian mitochondrial translation requires fidelity mediated by proofreading function of threonyl-tRNA synthetase

In a study published in PNAS, the team led by Prof. Zhou Xiaolong and Prof. Wang Enduo from the Center for Excellence in Molecular Cell Science, Shanghai Institute of Biochemistry and Cell Biology of the Chinese Academy of Sciences report that mammalian mitochondrial translation requires a high-level of accuracy at Thr codons, which is mediated by the proofreading (editing) function of mitochondrial threonyl-tRNA synthetase (mtThrRS).
Mitochondrion is one of the most crucial organelles in eukaryotes, regulating multiple cellular functions, such as energy production, redox homeostasis, calcium uptake and extrusion, and cell fate. Mammalian mitochondrion harbors its own genome (mtDNA), encoding 22 tRNAs, two rRNAs and 13 proteins, which are all essential subunits of respiratory complexes.
Aminoacyl-tRNA synthetases (aaRSs) ensure both speed and accuracy of mRNA translation by catalyzing tRNA aminoacylation. About half of aaRSs evolve an additional editing function to clear mischarged tRNAs to avoid mRNA mistranslation. Loss of editing of cytoplasmic aaRSs causes neurodegeneration.
However, most mitochondrial aaRSs have lost editing domains during evolution. Only four mitochondrial aaRSs (including mtThrRS) retain an intact editing domain. Whether such a simple mitochondrial translation with only 3,789 codons needs translational quality control remains unclear.
In this study, the researchers found that mtThrRS possesses editing capability to hydrolyze mischarged Ser-tRNAThr in vitro. They constructed a NIH3T3 cell line with simultaneous mutations (H138A/H142A) at the editing active site of mtThrRS, and revealed that larger abundance of Thr-to-Ser misincorporation was identified in the mtDNA-encoded proteins, demonstrating that mammalian cells critically rely on the editing function of mtThrRS in vivo.
Moreover, the researchers found that the mutant NIH3T3 cells exhibited impaired mitochondrial translation and oxidative phosphorylation, triggering oxidative stress and blocking the cell cycle in the G0/G1 phase. Remarkably, reactive oxygen species (ROS) scavenging by N-acetylcysteine treatment attenuated the abnormal cell proliferation.
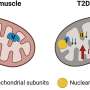
The researchers also established a heart tissue-specific mouse model with defective mtThrRS editing. They observed increased ROS levels, blocked cardiomyocyte proliferation, contractile dysfunction, dilated cardiomyopathy and cardiac fibrosis in the mutant mice.
This study elucidated that correct amino acid incorporation, mediated by mitochondrial aaRSs during mitochondrial gene expression, is essential for the structure and function of the organelle.
More information: Wen-Qiang Zheng et al, Mammalian mitochondrial translation infidelity leads to oxidative stress–induced cell cycle arrest and cardiomyopathy, Proceedings of the National Academy of Sciences (2023). DOI: 10.1073/pnas.2309714120
Journal information: Proceedings of the National Academy of Sciences
Provided by Chinese Academy of Sciences